Eric Faulques,a Florian Massuyeau,a Nataliya Kalashnykb and Dale L. Perryc
aInstitut des Matériaux Jean Rouxel, Université de Nantes, CNRS, UMR 6502, 2 rue de la Houssinière, BP 32229, F-4432 Nantes, France. E-mail: [email protected]
bInstitut des Sciences Moléculaires d’Orsay (ISMO), UMR 8214, CNRS and Université Paris-Sud, F-91405 Orsay Cedex, France
cMail Stop 70A1150, Lawrence Berkeley National Laboratory, University of California, Berkeley, CA 94720, USA
Introduction
Of all the elements in the Periodic Table, perhaps none is more complex than uranium.1,2,3 Because of its four oxidation states (+3, 4, 5 and 6) and the large sizes of its ions, uranium forms compounds and complexes that exhibit large coordination numbers, the number of atoms attached to the central uranium ion. Structurally documented uranium compounds offer many opportunities for study, since they exhibit exact bond angles, distances and crystal lattice details, allowing for theoretical studies involving electron density, electronic transitions and other calculations; some uranium compounds shown as minerals can be seen in Figure 1. These details also provide substantive bonding data for the development of more advanced studies of applied spectroscopy for analytical applications. The new detection approaches include the determination of ultra-trace levels of uranium by newly developed techniques, several using lasers.
 yellowish-green saleeite Mg(UO2)2(PO4)2•10H2O and (bottom panel) green metatorbernite Cu(UO2)2(PO4)2•8H2O. Legend: U (blue), O (red), P (violet), Cu (orange), Mg (green) and H (white). The saleeite photograph is reproduced by permission of Jolyon Ralph, C.J. Stefano and www.mindat.org.")
Uranium-based minerals afford especially excellent materials for a rigorous look at uranium and its chemistry and transport in the environment. Uranium minerals exhibit themselves as solid-state phases that possess crystal structures combining uranium with other elements such as lead, calcium, vanadium, titanium, copper, rare earth elements and aluminium, along with chemical functional groups such as phosphates, carbonates and silicates. They offer an array of analytically interfering elements and ions for the development of analytical techniques for looking at uranium under real environmental circumstances (rocks, soils, sands etc.), including the possibility of combined, multiple uranium mineral phases. An additional factor to realise is that these uranium solid phases—along with accompanying anions—control the amounts of uranium that undergo dissolution and undergo groundwater transport.
Raman scattering spectra
Raman scattering has extensively been used to study uranium compounds, both as laboratory molecular entities4,5,6 and as minerals.7, 8 Uranium compounds and minerals studied by Raman spectroscopy range from simple oxides to more complex species such as minerals that are discussed here. Many of the more complex uranium minerals contain the polyatomic core cation, the dioxouranium(VI), or uranyl ion. Other atoms (oxygen, sulfur, nitrogen etc.) and polyatomic anions (carbonates, phosphates etc.) are attached, or coordinated, to the central cation in complex structures found in minerals. Uranium minerals exhibit a large range of polyhedron geometries, bond-oxidation state parameters and polymerisation of polyhedra9 as part of their structures. All of the atoms in the molecule comprising the mineral structural lattice have, at some level, an impact on the Raman scattering spectra of the minerals.
While in theory, the uranyl ion (UO22+) is linear with symmetry D∞h, in fact, it is shown in the analysed mineral compounds studied here to exhibit a slightly bent (usually a few degrees less than 180°)C2v symmetry, assuming equal bond lengths, the same as the V-shaped water molecule. The representation of vibrations for the isolated bent uranyl ion in these minerals is Gvib=2A1+B1; therefore, all modes are active in the infrared (IR) and Raman spectra. The A1 modes represent the U–O symmetric stretching and the O–U–O bending vibrations with frequencies n1 and n2, respectively, and the B1 mode the antisymmetric U–O stretching vibration with frequency n3.
The ideally isolated PO43– ion has a tetrahedral structure with point group symmetry Td. The vibrations of this ion are represented by Gvib=A1+E+2T2, and all these modes are Raman active, while only the T2 modes are IR active in the free state. The symmetric and antisymmetric P–O stretching modes A1 and T2 occur at frequencies n1 and n3, respectively. The bending and deformation modes E and T2 occur at frequencies n2 and n4, respectively.
Note that the above assignments for the two isolated ions do not take into account the lattice environment in the minerals, the U–O and P–O bond length differences and the ion-site symmetry specific to each crystal. The latter can cause activation of vibrations in the infrared (IR) and splitting of doubly or triply degenerate modes. Nevertheless, this simplification helps in the recognition of the uranyl minerals.
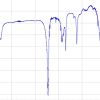
 micro-Raman spectra of several uranium phosphate minerals. (Right panel) atomic motions associated with the vibrations of the uranyl and phosphate ions. The U atom is six-coordinate in the structures. Here, we have represented motions of non-equatorial (eq) O atoms (Ot), but it is expected that the Raman system A can be assigned to both U–Oeq and U–Ot vibrations. The minerals noted in the figure are metatorbernite [(Cu(UO2PO4)2•8H2O], uranocircite [Ba(UO2)2(PO4)2•10-12H2O], saleeite [Mg[UO2PO4]2•10H2O], ulrichite [CaCu(UO2)(PO4)2•4H2O] and phosphuranylite [KCa(H3O)3(UO2)7(PO4)4O4•8H2O]. Spectra were acquired under a microscope with 100× objective magnification. The spot analysed is ≤2 µm, and the laser power on it is ≤0.5 mW.")
Figure 2 shows typical Raman spectra recorded with the Kr+ laser excitation line at 647.1nm for several uranyl phosphate minerals between 600cm–1 and 1200cm–1 in which the Raman bands can be used as markers to identify the mineral phases. Two distinctive sets of intense lines appear in the spectra, one around 800cm–1 (A) and another one (C) grouped around ~1000cm–1, 1020–1050cm–1 and 1100cm–1 (weak). Another very weak intensity region labelled B occurs around 900cm–1. These three band sets represent composite Raman frequencies of the pseudo-linear UO22+ central cations and the phosphate (PO43–) anions in the mineral structures. They can be assigned to the symmetric (n1) U–O stretching (A-system of lines, between 813cm–1 and 838cm–1) and to both symmetric and antisymmetric stretching vibrations (n1 and n3, respectively) of the P–O bonds (C-system of lines between 980cm–1 and 1100cm–1). The antisymmetric U–O stretching n3 of the uranyl ion should appear at higher wavenumber than the symmetric one. In Raman scattering, the n3(U–O mode) has a weak intensity and is only observed for uranocircite and metatorbernite at about 900cm–1 (B-system of lines). The line at 1100cm–1 is also very weak, and it can be assigned to the antisymmetric P–O stretching mode n3. The relative positions in the A and C systems (characteristic of n1 for UO22+ and n1, n3 for PO43–) vary considerably with the coordination sphere details of the uranyl cation. The multiple line intensities and their ratios to one another also change as a function of the different bonding structures and geometries exhibited in the crystal lattices.
The Raman lines of metatorbernite are completely confirmed by previous Raman studies on natural and synthetic metatorbernite undertaken with laser excitation lines at 633nm and 532nm, respectively.10,11 In the latter reference, the authors reported Raman peaks at 806cm–1 (shoulder), 827cm–1, 831cm–1 (shoulder), 905cm–1 (weak), 986cm–1 (shoulder), 997cm–1 and 1014cm–1 (weak), whereas we locate corresponding A-B-C peaks at 826cm–1,838cm–1 (shoulder), 904cm–1 (weak), 990cm–1, 995cm–1 and 1006cm–1.In Figure 2, the Raman spectrum of phosphuranylite (lines at 813cm–1, 827cm–1, 840cm–1, 997cm–1, 1008cm–1, 1050cm–1 and 1120cm–1) is almost the same as that of the Ruggles Mines sample described by Frost et al.12 using the 633nm excitation laser line which shows Raman peaks at 812cm–1, 832cm–1 (shoulder), 844cm–1, 1005cm–1, 1032cm–1, 1050cm–1 and 1125cm–1. However, it is slightly different from that described by Driscoll et al.13 for the 785nm excitation line. The RRUFF™ database14 gives Raman spectra of two 780 nm excited samples of meta-uranocircite, which has the same approximate chemical composition as uranocircite. The RRUFF spectrum of Sample I matches very well the Raman lines we find at 805cm–1 (shoulder), 823cm–1, 854cm–1, 988–1000cm–1 and 1111cm–1 for this mineral (appearing at 807cm–1, 824cm–1, 973cm–1, 997cm–1 and 1113cm–1 in that reference). The Raman spectrum of Sample II shows a richer number of lines (among them, the lines of interest for this study occur at 818cm–1, 823cm–1, 857cm–1, 995cm–1 and 1111cm–1), but the resemblance to our spectrum is unequivocal. Finally, the Raman spectra of ulrichite and saleeite have also been compiled in the RRUFF™ database for laser excitations 532nm and 780nm, and the line agreement with the spectra presented here is very good. The 780nm excited spectrum of ulrichite presents lines at 808cm–1, 979cm–1, 1025cm–1 and 1104cm–1 compared to lines located at 812–815cm–1 (broader), 980cm–1, 1025cm–1 and 1104cm–1 (weaker) in our study and with completely similar, relative intensities. For saleeite the A–C sets of lines are nearly identical, positioned at 838cm–1, 992cm–1 and 998cm–1 in Figure 2, and at 836cm–1, 913cm–1 and 990cm–1 in the RRUFF spectrum. These data also perfectly agree with another spectrum published for an excitation at 633nm.10 In general, the Raman scattering observed in the same uranyl phosphate mineralogical samples reported by different authors coincide very well in frequency and in relative intensity. On that account, it is remarkable that almost the same Raman signals can be found for each of these mineral species from different locations using different instrumentation and laser excitation lines.
As a result, our case study of micro-Raman spectroscopy applied to these five uranyl minerals demonstrates the reliability of micro-Raman spectroscopy which is able to identify complex, unique uranyl mineral phases with very similar chemical formulae (in the case of the present discussion, uranyl phosphate hydrates) but different crystallographic structures and lattices. Due to the very intense signal of the A-system in the spectra for all samples investigated, this technique is therefore suitable for precise tracking of uranyl moieties in contaminated soil, solid-state materials or even biological samples. Although this unambiguous Raman signature in the solid state does not require any further analytical techniques such as X-ray diffraction, in the next section, we present another spectroscopic probe which might be efficiently used for recognising uranyl phosphate and other anion-containing uranyl minerals.
Photoluminescence spectra
Photoluminescence (PL) spectroscopy can be run in many cases on Raman spectrometers with adapted laser excitations. This is a non-destructive method to explore the electronic structure of molecules and solids. In PL spectroscopy, monochromatic light is absorbed by the sample, a process referred to as photo-excitation, in which electrons are promoted to excited states by the excess energy brought to the system. The molecule or material dissipates this excess energy in its structure by electronic and geometric relaxation from the photo-excited states through decay by radiationless heat exchange and radiative re-combinations, producing emitted light called photoluminescence. The PL energy is the energy of the transition between the lowest excited state and the equilibrium state. PL spectroscopy covers wide, technical domains: measures of dopant and carrier concentration in silicon wafers, identification of defects in semiconductors, development of light emitting diodes and scintillators, biological tagging, super-resolved fluorescence microscopy of nano-objects, development of long lasting phosphors and other similar applications.
Uranium(VI), as found in the uranyl ion in the presently discussed minerals, possesses extraordinary photoluminescence properties with relatively long lifetimes ranging from several to hundreds of microseconds depending on its chemistry.15 In this respect, the emission characteristics of the uranyl cation are particularly attractive, owing to its broad distribution in geological and biological environments. Upon ultraviolet or blue light excitation, the emission of uranyl is imputable to an electronic transition from the lowest triplet excited state to the singlet ground state.16 This typifies strong fluorescence occurring in the green–yellow range of the visible spectrum, which exhibits, in most cases, a typical vibronic structure with energy gaps between the peaks from about 810cm–1 to 830cm–1 ± 10cm–1 (~0.1eV) corresponding to the symmetric stretching vibration of the uranyl ion in the different crystals. The vibronic structure is well developed at room temperature (RT) in four of the five minerals investigated in this study (see Figure 3). It is worth noticing that the peak energies of the fine structure vary with the mineral species, therefore providing a strong fingerprint for their identification in their native state. The most intense peak occurs at 519.5nm (19249 cm–1), 521.3nm (19183 cm–1), 525.6nm (19026 cm–1), 526.9nm (18979 cm–1) and 535.7nm (18667 cm–1) for saleeite, ulrichite, metatorbernite, uranocircite and phosphuranylite, respectively. Phosphuranylite presents a feature-less broad luminescence band at RT, which is also a signature of this species among the natural uranyl minerals analysed here.
 spectra of the uranium phosphate minerals excited with the 325 nm laser line. Spot size on the sample is ≤4 µm with a 40× objective magnification.")
The minerals differ in this spectroscopic experiment by their emission intensity and can be sorted by decreasing PL-intensity order as: uranocircite, metatorbernite, saleite, ulrichite, phosphuranylite. Ulrichite is not a very strong emitter and in phosphuranylite, the PL is relatively strongly quenched with respect to the three other compounds." />
Photoluminescence studies of the uranyl ion are extremely useful for investigating the chemical state of bonding of the ion in minerals, along with other studies of the role of both the uranyl ion and other uranium species in the environment. These include deactivation processes of uranium excited states in complexes that may occur from the uranium mineral dissolution,17 complexes of uranium in multiple oxidation states,18 investigation of uranium luminescence in a variety of solid state matrices,19 and quenching mechanisms and the role of possible intermediate uranium(V) species that can evolve from the uranyl ion in both solid state and liquid reactions.20
In summary, the two methods described here, Raman scattering and photoluminescence spectroscopy, are supplemental to each other when applied to uranium(VI) compounds that comprise uranium(VI) minerals. They yield mainly the vibrational and electronic energies, respectively, of the uranyl cation present in the minerals. Because the crystallographic structures are different from one mineral to another, one observes variations in the laser-induced spectra which are characteristic of the studied species. Laser-induced spectroscopy provides, therefore, powerful tools for direct speciation of uranium and identification of natural uranyl minerals relevant to the environment such as phosphate hydrates.
Acknowledgements
This work was partially supported by the US Department of Energy under Contract No. DE-ACO3-76SF00098.
References
- E.H.P. Cordfunke, The Chemistry of Uranium (Topics in Inorganic and General Chemistry). Elsevier Publishing Company, Amsterdam (1969).
- S. Cotton, Lanthanide and Actinide Chemistry (2nd Edition). Wiley, Chichester (2006).
- A.Y. Vasiliev and M. Sidorov (Eds), Uranium: Characteristics, Occurrence, and Human Exposure. Nova Science Publishers, Hauppauge, NY (2012).
- E. Faulques, R.E. Russo and D.L. Perry, “Raman spectral studies of uranyl sulfate and its urea complex structural isomers”, Spectrochim. Acta A 49, 975–983 (1993). doi: http://dx.doi.org/10.1016/0584-8539(93)80216-W
- E. Faulques, R.E. Russo and D.L. Perry, “Raman studies of uranyl nitrate and its hydroxy bridged dimer”, Spectrochim. Acta A 50, 757–763 (1994). doi: http://dx.doi.org/10.1016/0584-8539(94)80013-8
- T.R. Ravindran and A.K. Arora, “Study of Raman spectra of uranium using a surface-enhanced Raman scattering technique”, J. Raman Spectrosc. 42, 885–887 (2011). doi: http://dx.doi.org/10.1002/jrs.2926
- R.L. Frost, J. Cejka and M.L. Weier, “Raman spectroscopic study of the uranyl oxyhydroxide hydrates: becqerelite, billietite, curite, schoepite, and vandendriess-cheite”, J. Raman Spectrosc. 38, 460–466 (2007). doi: http://dx.doi.org/10.1002/jrs.1669
- G. Guimbretiere, A. Canizares, P. Simon, Y.A. Tobon-Correa, M.R. Ammar, C. Corbel and M.F. Barthe, “In-situ Raman observation of uranium dioxide weathering exposed to water radiolysis”, Spectrosc. Lett. 44, 570–573 (2011). doi: http://dx.doi.org/10.1080/00387010.2011.610857
- P.C. Burns, “U6+ minerals and inorganic compounds: insights into an expanded structural hierarchy of crystal structures”, Can. Mineral. 43, 1839–1894 (2005). doi: http://dx.doi.org/10.2113/gscanmin.43.6.1839
- R.L. Frost, “An infrared and Raman spectroscopic study of the uranyl micas”, Spectrochim. Acta A 60, 1469–1480 (2004). doi: http://dx.doi.org/10.1016/j.saa.2003.08.013
- N. Sanchez-Pastor, A.J. Pinto, J.M. Astilleros, L. Fernandez-Diaz and M.A. Gonçalves, “Raman spectroscopic characterization of a synthetic, non-stoichiometric Cu-Ba uranyl phosphate”, Spectrochim. Acta A 113, 196–202 (2013). doi: http://dx.doi.org/10.1016/j.saa.2013.03.094
- R.L. Frost, J. Cejka and G. Ayoko, “Raman spectroscopy study of the uranyl phosphate minerals phosphuranylite and yingjiangite”, J. Raman Spectrosc. 39, 495–502 (2008). doi: http://dx.doi.org/10.1002/jrs.1868
- R.J.P. Driscoll, D. Wolverson, J.M. Mitchels, J.M. Skelton, S.C. Parker, M. Molinari, I. Khan, D. Geeson and G.C. Allen, “A Raman spectroscopic study of uranyl minerals from Cornwall, UK”, RSC Adv. 4, 59137–59149 (2014). doi: http://dx.doi.org/10.1039/C4RA09361E
- http://rruff.info/Metauranocircite/R070721 and http://rruff.info/ Metauranocircite Ibidem /Saleeite; Ibidem/Ulrichite
- G. Meinrath, S. Lisc, Z. Strylad and C. Noubactep, “Lifetime and fluorescence quantum yield of uranium(VI) species in hydrolyzed solutions”, J. Alloy. Compd. 300–301, 107–112 (2000). doi: http://dx.doi.org/10.1016/S0925-8388(99)00739-2
- J.B.M. Novo, F.R. Batista, C.J. da Cunha, L.C. Dias, Jr, and F.B.T. Pessine, “Time-resolved studies in hydrogen uranyl phosphate intercalated with amines”, J. Lumin. 124, 133–139 (2007). doi: http://dx.doi.org/10.1016/j.jlumin.2006.02.009
- S.J. Formosinho, H.D. Burrows, M.Da Graca Miguel, M.E.D.G. Azenha, I.M. Saraiva, A.C.D.N. Ribeiro, I.V. Khudyakov, R.G. Gasanov, M. Bolte and M. Sarakha, “Deactivation processes of the lowest excited state of [UO2(H2O)5]2+ in aqueous solution”, Photochem. Photobiol. Sci. 2, 569–575 (2003). doi: http://dx.doi.org/10.1039/B300346A
- E. Hashem, J.A. Platts, F. Hartl, G. Lorusso I.M. Evangelisti, C. Schulzke and R.J. Baker, “Thiocyanate complexes of uranium in multiple oxidation states: a combined structural, magnetic, spectroscopic, spectroelectrochemical, and theoretical study”, Inorg. Chem. 53, 8624–8637 (2014). doi: http://dx.doi.org/10.1021/ic501236j
- M. Mohapatra, B. Rajeswari, R.M. Kadam, M. Kumar, T.K. Seshagiri, N.K. Porwal, S.V. Godbole and V. Natarajan, “Investigation of uranium luminescence in SrB4O7 matrix by time resolved photoluminescence, thermally stimulated luminescence and electron spin resonance spectroscopy”, J. Alloy. Compd. 611, 74–81 (2014). doi: http://dx.doi.org/10.1016/j.jallcom.2014.05.096
- S. Tsushima, C. Gotz and K.F. Dr, “Photoluminescence of uranium(VI): quenching mechanisms and role of uranium(V),” Chem. Eur. J. 16, 8029–8033 (2010). doi: http://dx.doi.org/10.1002/chem.201000408