Bernd W.K. Diehl,a Frank Malzb and Ulrike Holzgrabec,*
aSpectralservice, Emil-Hoffmann-Str. 33, 50996 Köln, Germany
bFederal Institute for Materials Research and Testing (BAM), Richard-Willstätter-Str. 11, 12489 Berlin, Germany
cInstitute of Pharmacy and Food Chemistry, University of Würzburg, Am Hubland, 97074 Würzburg, Germany
Introduction
Quantitative (q) methods in nuclear magnetic resonance (NMR) spectroscopy have been used successfully for many years. However, there is a lack of general acceptance of qNMR in the pharmaceutical industry, whereas chromatographic methods are well established and documented in analytical standards, pharmacopoeia and drug master files. However, NMR spectroscopy can be considered as a quantitative primary ratio method of measurement,1,2 since the ratio of substances in a mixture can be determined directly from the NMR spectral measurement without referencing to another substance. The absolute amount of substances can be determined by using simple reference substances.
The purpose of this short review article is to highlight some capabilities of qNMR spectroscopic methods in drug quality evaluation, indicating that qNMR spectroscopy should be more often applied when chromatographic methods are not working effectively.
NMR spectroscopy can be used in different fields of the quality evaluation of drugs:3
- to identify a drug
- to determine the level of impurities and elucidate their structure, and to observe the course of decomposition
- to evaluate the content of residual solvents
- to determine the isomeric composition: the ratio of diastereomers and the enantiomeric excess (ee) by means of chiral additives
- to determine molar ratios of (protonated) basic drugs and (deprotonated) organic acids in respective salts.
Basic qNMR
The most important fundamental relation for qNMR is that the signal response IX (signal area) in a spectrum is directly proportional to the number of nuclei NX generating the corresponding resonance line:
IX = kSNX
with kS being a spectrometer constant. The prerequisite for quantitative measurements is that kS remains constant for all resonance lines within a NMR spectrum. A number of criteria have to be fulfilled for ensuring this:
(1) The pulse excitation must be uniform for the entire spectral width (SW) of interest, which requires short pulses (typically 10 µs). In this case, 1H NMR spectra can be acquired quantitatively. However, spectra of heavier nuclei (19F, 31P etc.) with larger chemical shift ranges may suffer intensity distortion, particularly if measured at very high magnetic fields.4
(2) The repetition time τ (or recycling time) depends on the longest longitudinal relaxation time T1 of all signals of interest. The T1 relaxation is described by:
\[{M_z} = {M_0}\left( {1 - {e^{{\raise0.5ex\hbox{$\scriptstyle { - \tau }$} \kern-0.1em/\kern-0.15em \lower0.25ex\hbox{$\scriptstyle {{T_1}}$}}}}} \right)\]
with Mz and M0 being the magnetisation along the z-axis (response factor) after waiting time τ and at thermal equilibrium, respectively. Routinely, measurements are done by choosing τ = 5 × T1. Hence, 99.3% of the equilibrium magnetisation (signal) is measured.4 For nuclei with very long T1 values, e.g. 13C, 29Si, 31P etc., paramagnetic relaxation reagents such as chromium(III) acetylacetonate [Cr(acac)3] are added in order to shorten the relaxation times.
(3) Heteronuclear NMR experiments of X nuclei (13C, 19F, ...) with simultaneous 1H broadband decoupling cause inherent intensity distortions by the nuclear Overhauser effect (nOe). This nOe effect on the spectrometer constant kS can be described5 by:
\[{k_S} = {k_0} \cdot \left( {1 + \eta } \right) \cdot \left( {\frac{{1 - {e^{ - \frac{\tau }{{{T_1}}}}}}}{{1 - \cos \alpha \cdot {e^{ - \frac{\tau }{{{T_1}}}}}}}} \right) \cdot \sin \alpha \]
where k0 is a constant instrumental factor, η is the nuclear Overhauser enhancement factor and α is the flip angle of the excitation pulse. In order to suppress possible intensity distortions below 1%, the following rules must be fulfilled: (i) 1H decoupling is applied only during the signal acquisition time (inverse gated technique) in order to minimise η, (ii) the repetition must be set to 5 ... 7 × T1,6 and (iii) 90° pulse should be used for the excitation.
Additional parameters may affect the measurement uncertainty (viz. accuracy and precision) of the signal intensities in NMR spectra:
(4) The required acquisition time taq depends on the smallest line width in the spectrum, and truncation of the NMR signal in the time domain [free induction decay (FID)] must be avoided. If truncation occurs, signal forms with (substantial) “wiggles” appear in the spectra, and, in combination with FID baseline correction modes, wrong intensities will result.7 Usually, the signal should decay completely half way through the acquisition period. It also ensures that enough data points describe the NMR lines (at least five data points above the half-height) such that the integration procedure does not cause artificial distortions (intensity error less than 1%).8–10
(5) The accuracy of the integration procedure depends on the signal-to-noise-ratio (S/N). For a precise integration a maximum S/N is required. For a precision better than 1%, S/N of 250 : 1 (1H), 300 : 1 (19F) and 600 : 1 (31P) have been proposed in the literature.11 It should be noted that there is no reason for these different numbers; one and the same S/N should yield the same precision, irrespective of the NMR frequency (nucleus).
Nevertheless, a detailed validation of qNMR using single pulse excitation of 1H NMR spectra proved that the method is robust, if acquisition times, repetition times and the signal-to-noise-ratios are set according to rules mentioned above.2
Sample preparation
Easy and fast sample preparation is possible for qNMR purposes. For relative measurements, the analyte is dissolved and diluted in an appropriate deuterated solvent. In contrast, for absolute measurements, a one-point calibration has to be carried out by adding a standard of known assay gravimetrically such that the intensity ratio of the analyte signals of interest and the standard are nearly equally strong. Special requirements for the standards have been described elsewhere.12,13
Spectra evaluation (integration)
The operator is most commonly the main error source of qNMR measurements for various reasons. Baseline and phase correction must be performed with very high precision to ensure accurate results.7,14 It was found that automatic routines could not solve this problem in general; therefore the operator’s experience determines the quality of the results.2 Furthermore, the integration limits for the NMR lines have to be set according to the full width at half height (FWHH) of each signal. Extending the integration over a frequency range of 64 times the FWHH value ensures that 99% of the entire signal intensity (Lorentzian) is obtained.
Quantitative NMR spectroscopy
Relative method
The determination of ratios is the easiest way to obtain quantitative results by NMR. The molar ratio nX/nY of two compounds X and Y can be calculated straightforwardly using:
\[\frac{{{n_{\rm{X}}}}}{{{n_{\rm{Y}}}}} = \frac{{{I_{\rm{X}}}}}{{{I_{\rm{Y}}}}} \cdot \frac{{{N_{\rm{Y}}}}}{{{N_{\rm{X}}}}}\]
In this relation kS cancels provided it is constant (a necessary prerequisite, see above) for all lines. Consequently, the amount fraction of a compound X in a mixture of m components is given by:
\[\frac{{{n_{\rm{X}}}}}{{\sum\limits_{i = 1}^m {{n_i}} }} = \frac{{I{}_{\rm{X}}/{N_{\rm{X}}}}}{{\sum\limits_{i = 1}^m {{I_i}/{N_i}} }} \cdot 100\% \]
irrespective of the solvent signal in which the mixture is dissolved.
Furthermore, qNMR is a most important method for quantifying the ratios of isomers, diastereomers and enantiomers,15 because of its excellent selectivity for structural analysis combined with the easy and fast quantification procedure. Even the knowledge of the molar masses of the components is not required. It should be noted, that enantiomers, although having identical spectra, can be differentiated by NMR too, if chiral solvents or complexing agents16,17 are used.
Absolute method
qNMR offers two different methods for quantifying absolute values such as content concentration or assay.
(i) If all impurities show up in the NMR spectrum, and if they can be assigned structurally and if they can be measured quantitatively, the assay is simply the difference from the 100% value (so called 100% method). This approach is limited for partially overlapping signals, and it is impossible for impurities not containing the observed nucleus [e.g. inorganic impurities (NaCl) in case of 1H NMR].
(ii) The main component PX can be calculated directly from the NMR using a standard of known assay PStd
\[{P_X} = \frac{{{I_{\rm{X}}}}}{{{I_{{\rm{Std}}}}}} \cdot \frac{{{N_{{\rm{Std}}}}}}{{{N_{\rm{X}}}}} \cdot \frac{{{M_{\rm{X}}}}}{{{M_{{\rm{Std}}}}}} \cdot \frac{{{m_{{\rm{Std}}}}}}{m} \cdot {P_{{\rm{Std}}}}\]
with MX and MStd being the molar masses of analyte and standard, m and mStd the weights of the sample and standard, and PX and PStd the assays of analyte and standard, respectively.
Furthermore, other procedures for quantifying concentrations or assays exist using NMR. The use of an internal standard is the most popular. If contamination of the analyte by the chosen standard must be avoided, external standards have to be used. This can be achieved either by putting a capillary, filled with the dissolved standard, inside the analyte NMR tube or by using two NMR tubes one filled with the analyte solution, the other filled with the standard solution. In both cases analyte and standard should be dissolved in the same solvent and the volumes of the tubes (capillary) have to be calibrated before. Capillaries are commonly used for the determination of deuterated assays or the water content of solvents. The standard addition method is a third possibility of absolute value determinations. If known amounts of the active compound are added to the solution in several steps the content can be calculated without the knowledge of the molar mass of the analyte.
In 1998 Akoka and co-workers18,19 published a new technique called ERETIC (Electronic Reference To access In vivo Concentrations). Here, an electronic reference signal is fed in the resonance circuit of the probe during the acquisition time using a free coil in the probe (heteronuclear channel). Amplitude, FWHH and frequency (chemical shift position) can be set by the operator. The advantage is that this electronic signal can be shifted to free spectral range avoiding superposition with the analyte signals.
Selectivity
The analyte NMR signal used for the quantification procedure must be assigned unambiguously in terms of the molecular structure (molecular groups). It must be ensured that this monitor signal belongs exclusively to the analyte without any contributions from an impurity. If it nevertheless happens, one can (i) change the temperature, (ii) the pH value, (iii) add specific shift reagents, (iv) use another solvent or different concentration in order to separate such overlapping signals.
If these attempts fail, one may search for another (resolved) signal of the same impurity. If the specific impurity resonances can be assigned unambiguously and determined quantitatively, the hidden impurity signal can be subtracted from the total signal area.
Another serious problem may arise if the impurity signals cannot by distinguished from the analyte at all. This situation was met in a purity determination of atrazine containing simazine and propazine as impurities.20 The authors used elevated temperatures and homodecoupling to tackle the problem.
Applications in international pharmacopoeias
Due to the broad range of possibilities for application,21 international pharmacopoeias such as the European Pharmacopoeia (EP, PhEur 6th edition), the British Pharmacopoeia (BP) and the United States Pharmacopeia USP29, cite use of NMR spectroscopy for identification purposes and quantitative NMR spectroscopy for evaluation of the composition of polymers, mainly excipients and impurities in drugs. The PhEur 6th edition describes the method of NMR spectroscopy only in principle using continuous wave (no longer in use nowadays) and pulsed spectrometry. In contrast, beside the description of the physical background of NMR spectroscopy, the apparatus, the general method and the interpretation of a spectrum, the USP 29 gives detailed information about procedures to be applied for both qualitative and quantitative purposes. For quantitative applications an absolute method utilising an internal standard and a relative method is given.
A special working group on NMR spectroscopy in the European Directorate for Quality of Medicine has been commissioned which will adopt the general monograph on NMR spectroscopy in the EP to modern applications utilising 1D and 2D methods, different methods of quantification, and liquid and solid phase measurements.
Currently, the following applications can be found in the three aforementioned pharmacopoeias.
Identity
PhEur 5.4: Buserelin (1H), Goserelin (13C), Tobramycin (1H); BP 1998: Hydrocortisone Sodium Phosphate (1H); USP29: Amylnitrite isomers (1H); PhEur 5.4: Heparins low-molecular-mass (13C), vaccines such as Haemophilus Type b conjugate vaccine, Meningococcal Group C Conjugate Vaccine and Pneumococcal Polysaccharide Conjugate Vaccine (adsorbed) (1H), and Salmon oil farmed {distribution of [b(2)-acyl] of fatty acids (13C)}.
Tests
DAB 9: Gentamicin (1H); PhEur 5.4: Poloxamer ratio of oxypropylene/oxyethylene; Hydroxypropylbetadex: molar substitution (1H); USP29: Orphenadrine citrate: meta/para isomer (1H).
Assay
USP29: Amylnitrite isomers (1H).
For identification purposes the pharmacopoeias use NMR spectroscopy in a similar way to IR spectroscopy, i.e. by a comparison of the spectra of the chemical reference substance and the substance being examined. Thus, NMR spectroscopy is mainly applied to complex molecules such as, for example, hydrocortisone, goserelin, buserelin, tobramycin and different low-molecular mass heparins such as dalteparin, enoxaparin, parnaparin, certoparin and nadroparin. Recently, 1H NMR was introduced for characterisation of conjugated vaccines in the EP in addition to the classical methods.
Drug and active pharmaceutical ingredient analysis by qNMR
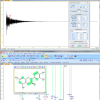
Drug analysis is probably the most important application for qNMR. In one step it is possible to measure several parameters for drug acceptance or storage stability. NMR spectroscopy provides both identification and quantification of the active ingredient, of by-products and degradation products, of excipients and solvent residues. Application of qNMR for characterisation of reference compounds derived from natural materials is well established; their reliability was confirmed in round-robin tests.22 Several examples for drug analysis are reported in the literature, e.g. the quantification of hydroquinone in wound glue,23 which is complicated for chromatographic methods due to the reactivity of the acrylate system. In this short article the potential of qNMR analysis will be illustrated on the basis of only a few simple examples. For the analysis of a medical formulation containing a platinum complex the internal standard 3-(3,4-dihydroxyphenyl)-l-alanin (DHPA) was used. The oxaliplatin as well as the formulation aid lactose can be quantified in an aqueous solution containing added D2O as the reference signal (lock at 0 ppm) (see Figure 1).
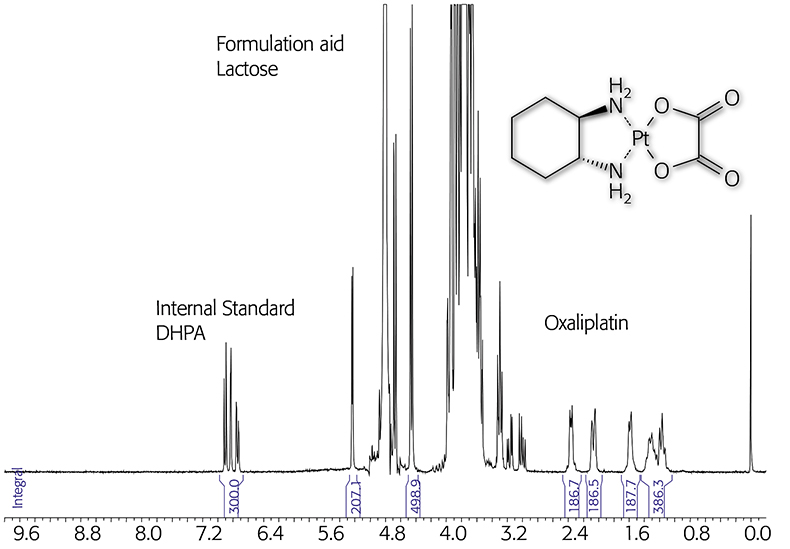
Figure 1. qNMR of an oxaliplatin formulation using the internal standard DHPA (300MHz).
Hetero nuclei containing drugs are also targets for qNMR. Phosphorous containing molecules such as chlodronic or ethidronic acid have been analysed by routine qNMR for many years.24 Quantification and simultaneous determination of enantiomeric excess in the case of cyclophosphamide and related substances using different cyclodextrin species have been reported.23 Many drugs and pesticides contain the hetero nuclei fluorine or phosphorous atoms. A 100% natural abundance of 19F and 31P predestines NMR spectroscopy as the selective and sensitive analytical tool for quantification of these chemicals, even in complex matrices. The only requirement is the availability of a suitable internal standard and probably also a valid extraction method. Hetero nuclear qNMR is an especially powerful tool in the analysis of by-products in amounts of 0.1% and less. The structural similarity of active compounds and their by-products results in an almost identical relaxation behaviour; thus, by knowledge of the respective molecular structures no specific standard is necessary for quantification.
Phytopharmaceutics are another domain of qNMR applications. For example, the NMR spectra of Aloe vera inner gel (GEL) and Aloe vera whole leaf (WL) are discussed as examples for qualitative and quantitative analysis to distinguish between different plant sources and different qualities of extracts. For economical reasons it is necessary to characterise whether a product was taken from the inner gel of Aloe vera, which is more expensive than products from the whole leaf. The composition of fruit acids derived from the citric acid cycle can be used as a marker to distinguish between raw materials. Only the green part of Aloe vera produces citric and isocitric acid, the latter is partly converted to the lactone. If both molecules can be detected in an Aloe vera product, the sample is therefore either a whole leaf material or it is contaminated with parts of the cortex.
Using the internal standard method with nicotinis acid amide (NSA) the characterisation of each Aloe vera sample can be performed quantitatively or at least semi-quantitatively. Malic acid as well as glucose and the polymer Aloverose are the main constituents of fresh Aloe vera gel and are quantified routinely by qNMR. Additional signals originatinge from WL material are quantified the same way (Figure 2).
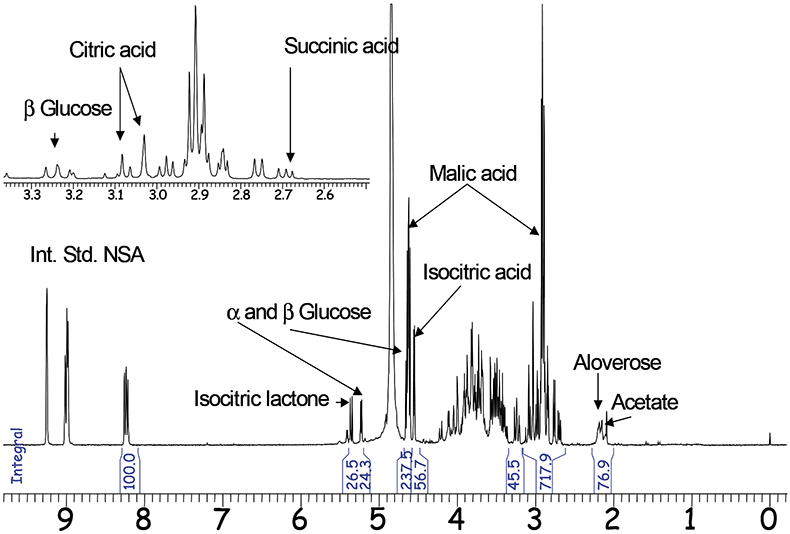
Figure 2. 1H-NMR spectrum of Aloe vera whole leaf (300MHz).
In addition to the main constituents chemical and bacterial degradation products, additives (e.g. preservatives) and adulterating agents can be quantified in the same qNMR analysis.24
Excipients
Besides the active molecules in drugs, many excipients, including polymers, can be analysed qualitatively and often quantitatively. Emulsifiers are often a mixture of similar molecules (isomers or homologues, e.g. para-isononylphenol, fatty acid mixtures). By reaction of these basic materials (alcohols, amines, fatty acids, triglycerides, carbohydrates, phenols etc.) with ethylene oxide and/or propylene oxide, polymeric structures are formed with a wide variety of molecular weights. The quantitation of emulsifiers by qNMR based on the ratio of oxyethylene and oxypropylene containing copolymers is described in EP5 and USP29 (see above). A similar procedure is used for characterisation of polylactid/polyglycolid23 copolymers. Furthermore, 13C qNMR enables the determination of polyoxyethylene chain length in PEG or modified PEG ab initio by comparison of the integral areas of the terminal methylene groups with the sum of the methylene chain.24
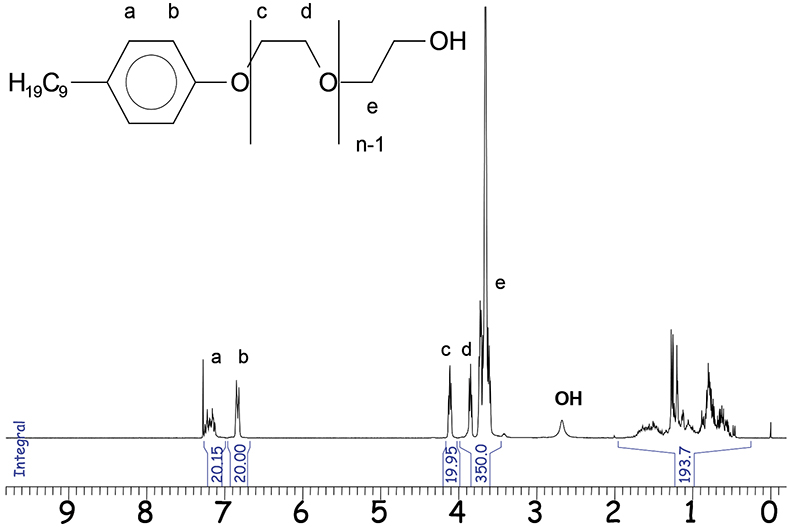
Figure 3. 1H-NMR spectrum of NPE (3000MHz).
One representative 1H-NMR application is the analysis of nonylphenol polyethylene glycol (NPE). The functional groups of these mixtures cluster to separated signal areas in an NMR spectrum as shown in Figure 3.
By normalisation of the integral areas to 10 for a single, proton the mean ethylene glycol (EO) distribution and the alkyl chain lengths are directly determinable. The mean number n of EO units (–CH2–CH2–O–) in the respective sample is:
n = (19.95 + 350)/40 = 9.25
The integral value of the alkyl chain between δ = 0.5 and 2.0 ppm confirms the presence of 19 protons in the alkyl part. Quantification of NPE from complex formulations is possible by standard addition or using the specific integrals at δ = 6.8 ppm (b) or at δ = 4.1 ppm (c). However, for calculation the aromatic protons at δ = 6.8 ppm are most suitable. Beside NPE, there are more complex emulsifiers based on alkylated phenols. Differences in their molecular structure can be identified by NMR spectroscopy. The terminal parts of the respective emulsifier molecules, e.g. the aromatic protons enable the calculation of the polyethylene and polypropylene glycol (PO) chain lengths and the mean molecular weight.
Through these few examples the value and versatility of qNMR spectroscopy to the quality and compositional analyses of active pharmaceutical ingredients and organic excipients has been clearly demonstrated, and it is expected that its use in these areas of application will continue to increase significantly.
References
- T.J. Quinn, Metrologia 34, 61–65 (1997).
- F. Malz and H. Jancke, J. Pharm. Biomed. Anal. 38, 813–823 (2005).
- U. Holzgrabe, R. Deubner, C. Schollmayer and B. Waibel, J. Pharm. Biomed. Anal. 38, 806–812 (2005).
- R. Freeman, Handbook of Nuclear Magnetic Resonance. Addison Wesley Longman, Edinburgh (1997).
- F. El-Shahed, K. Doerffel and R. Radeglia, J. Prakt. Chem. 321, 859–864 (1979).
- J.R. Mooney, in Analytical NMR, Ed by L.D. Field and S. Sternhell. John Wiley & Sons, Chichester (1989).
- D.L. Rabenstein, K.K. Millis and E.J. Strauss, Anal. Chem. 60, 1380A–1391A (1988).
- F.G. Herring and P.S. Philips, J. Magn. Reson. 62, 19 (1985).
- J.P. Grivet, Signal Treatment and Signal Analysis in NMR. Elsevier, Amsterdam (1996).
- K. McLeod and M.B. Comisarow, J. Magn. Reson. 84, 490–500 (1989).
- T.S. Al Deen, Dissertation, University of South Wales, Sydney, Australia.
- L. Griffiths and A.M. Irving, Analyst 123, 1061–1068 (1998).
- C.K. Larive, D. Jayawickrama and L. Orfi, Appl. Spectosc. 51, 1531–1536 (1997).
- D.L. Rabenstein and D.A. Keire, Pract. Spectrosc. 11, 323–369 (1991).
- R. Deubner and U. Holzgrabe, Magn. Reson. Chem. 40, 762–766 (2002).
- G.M. Hanna, Enantiomer 5, 303–312 (2000).
- J.S. Salsbury and P.K. Isbester, Magn. Reson. Chem. 43, 910 (2005).
- S. Akoka, S. Barantin and M. Trierweiler, Anal. Chem. 71, 2554–2557 (1999).
- G.S. Remaud, V. Silvestrs and S. Akoka, Accred. Qual. Assus. 10, 415–420 (2005).
- F. Malz and H. Jancke, Anal. Bioanal. Chem. 385, 760–765 (2006).
- U. Holzgrabe, R. Deubner, C. Schollmayer and B. Waibel, J. Pharm. Biomed. Anal. 38, 806–812 (2005).
- B.W.K. Diehl, Round-robin test, unpublished results.
- U. Holzgrabe, I. Wawer and B. Diehl, NMR Spectroscopy in Drug Development and Analysis. Wiley-VCH, Weinheim (1999).
- Spectral Service, information available.