Antony N. Daviesa,b and Roy Goodacrec
and Roy Goodacrec
aExpert Capability Group – Measurement and Analytical Science, Nouryon, Deventer, the Netherlands
bSERC, Sustainable Environment Research Centre, Faculty of Computing Engineering and Science, University of South Wales, UK
cDepartment of Biochemistry, Institute of Integrative Biology, University of Liverpool, UK.
E-mail: [email protected]
During the recent Royal Society of Chemistry, Faraday discussion meeting in Edinburgh on Challenges in Analysis of Complex Natural Mixtures I found myself wondering if the power that our modern spectrometers bring to the study of highly complex systems can sometimes overwhelm our natural scepticism around poor sampling practices.1 Some targeted questions put by Roy Goodacre in this direction to several speakers seemed to indicate I was not alone in my concerns, so I thought it might be worth looking at the temptations and some good practices in this area.
My spectrometer has identified 30,000 separate chemical entities so why do I need eight replicate samples?
As regular readers know, this column never aims to be deliberately provocative (!) but as our analytical spectroscopic and spectrometric toolbox gets stronger and stronger, there is always going to be a temptation to revel in the glory of the latest high-resolution enhancement for its own sake and to forget, just for a moment, why we are carrying out the experiments in the first place. In the world of omics experiments it is even more important to be sure that the results we are churning out by the Petabyte are robust and fit-for-purpose. If we leave aside the cost of the instrumentation, the societal costs of sloppyomics as more data becomes openly available for other scientists to use, could lead to false conclusions being drawn and resources being diverted down apparently promising dead-ends. We are reminded by George Poste in his editorial “Bring on the Biomarkers” in 20112 that, at that time, of the 150,000 clinical biomarkers described in the literature a mere 100 were routinely used in the clinic.
Omics experiments in themselves present an enormous issue for classical statisticians just by their huge dimensionality. Conventional wisdom has it that the greater the dimensionality of your problem, the greater the number of unique un-related samples you need if you wish to analyse the problem successfully. But where the promises of the omics approach are being sung the loudest is also the area where it is always notoriously difficult to recruit large sample populations.
In the health care environment, omics is believed to be one of the key analytical spectroscopic advances which will form the backbone of personalised medicine. However, inconsistent ethics committees, medical practitioner patient notes and a simple lack of enough patients taking part in trials who are the same sex, age, weight (or BMI), ethnic origin, diet, alcohol intake, fitness regime, medical history etc. and who are, for example, at the same stage of say a non-small cell carcinoma, could well hinder this approach well into the future. Let us not even start discussing the need for healthy controls with the same characteristics or even wander into the analytical minefield of comparing results from continuous monitoring against grab sampling with different storage strategies taken by different projects. Let us not forget that in most case-control studies the cases (those with some form of disease) are usually already on medication (or self medicating), so this strong confounding factor also needs to be considered.
There is an enormous gap between delivering theoretical correlations with the hope of finding causation from studies of cell cultures in Petri dishes to catching the developing lung cancer in a fit, football-playing 45 year-old engineer before he starts coughing blood into his handkerchief.
So is it really appropriate in such an environment to ignore all our Good Sampling Practice that was drummed into us at university (hopefully) and just go all out for as much data as we can get and (ethics committees willing) just keep throwing the mass spectra, nuclear magnetic resonance (NMR) data sets and our ion mobility fingerprints onto a big pile for the statisticians to fight over? Several years ago Raji Balasubramanian and co-workers compared some classification algorithms used in omics spectroscopic technologies driven by the high-dimensionality of the data.3 Lauren McIntyre looked last year at the lack of samples compared to the complexity of metabolites/genes and the lack of acknowledgement of over-fitting in the literature proposing a slightly different two-stage approach to the data analysis challenge.4 Drupad Trivedi and colleagues recently surveyed the metabolomics literature and found that the vast majority of studies were unfortunately underpowered.5 At the beginning of this year, Wu and co-authors published a “selective” review on integrating data from different types of omics experiments who want to add another level of complexity to their lives!6 Thus an absolutely essential components of any study that generates megavariate data is the need to reduce false discovery.7
If so, then how on earth do we continue to convince the governmental funding bodies that it is wise to pour money into these areas of research in the long term? Those in the medical spectroscopy field who passionately believe in this approach, will need to answer the question every three to five years about how many lives did your last project save? (As those approaching the next UK Research Excellence Framework will have to think about…). Maybe the best approach is to keep all these issues in mind when designing your experiments in the first place as the next story shows.
Studying the after effects of a natural disaster by omics
Tohoku Medical Megabank (TMM) Project was created to operate prospective cohort studies in Japan for regions where the population were impacted by the Great East Japan Earthquake on 11 March 2011.8 The project has at its heart the desire to support personalised medical support for the earthquake-damaged regions in the future. A good deal of thought went into this multi-omics study going right back to the sampling procedures. Two cohort studies are discussed—one an adult study and a second birth and three generations study with over 150,000 participants being recruited from 2013 to 2017. Molecular profiling of each participant is important to catch genetic and environmental factors. The analytical centre at the biobank carries out standard non-targeted mass spectrometry (MS) and NMR analyses making the data available to the scientific community.
The authors discussed the difficulty in carrying out sample collection during omics cohort studies—where although the genome will not alter, target metabolites may well be unstable and will be influenced by many factors which must be captured at the time of sampling. Indeed, they make the nice statement that the quality of the omics data largely depends on the quality of the collected samples. They studied which type of blood collection procedure was best for omics studies, deciding that it was best to collect EDTA plasma, as proteins and metabolites can be unstable during serum clotting. To remain consistent with other laboratories, however, they decided to continue to collect both serum and plasma samples. Figure 1 shows the sample collection and transportation plan from the cohort recruitment sites to the biobank.
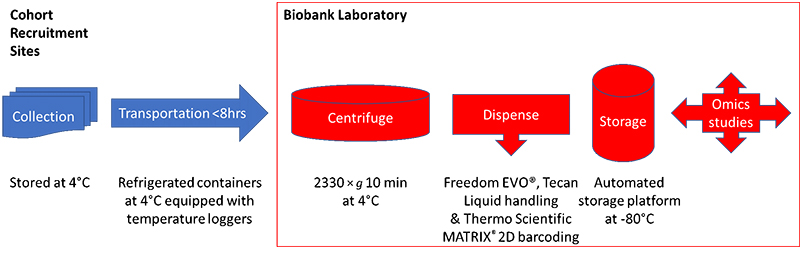
Figure 1. The TMM cohort sample collection and storage protocol.
The TMM central laboratory protocols for proteome and metabolome analysis
For proteome analysis, the TMM team carried out LC-MS/MS measurements in triplicate of the plasma samples after they had been denatured, reduced and alkylated followed by digestion and de-salting. Unfortunately, these studies take over an hour per sample, which clearly was going to cause problems with a project of this size.
For the metabolome analysis they adopted a non-targeted approach using NMR and both positive- and negative-ion mode LC-MS.
Metabolites were extracted from the plasma samples into a sodium phosphate buffer for the NMR studies on a 600 MHz instrument collecting standard 1D NOESY and CMPG spectra successfully identifying and quantifying 37 metabolites. For the MS analyses, an automated sample preparation robot was used which could process around 100 samples per hour, detecting over 1000 peaks and identifying 250 metabolites. For positive-ion mode analyses, the team used an UHPLC QTOF/MS system with electrospray ionisation and a C18 column (Acquity HSS T3; Waters) was used for LC separation. For negative-ion mode, a NANOSPACE SI II HPLC (Shiseido, Tokyo) and a Q Exactive Orbitrap system was deployed using a HILIC column for separation (ZIC pHILIC; Sequant, Darmstadt). The MS measurements could be run at four per hour.
Sample quality control in metabolome analysis
Not satisfied with the level of sampling standardisation and analysis described above, the team also put protocols in place on the not unreasonable assumption that there would be some sample handling errors dealing with such a large study. Samples were excluded as outliers if the NMR data on certain control metabolites were outside the ranges expected. For example, the blood glucose values needed to be no lower than 70 % of that measured by an original blood test carried out at the recruitment site and the lactose levels could not exceed more than 2× standard deviation of the cohort average. Samples were also excluded if they breached some aspect of the protocol, such as accidental storage for longer periods before entering the biobank.
Finally, the quality controlled data are being made available at the jMorp Japanese Multi Omics Reference Panel at https://jmorp.megabank.tohoku.ac.jp/201905/ and 8 May saw jMorp release 201905 of the 5KJPNv2 Genotype Frequency dataset from 3500 individuals. The metabolites database release (ToMMo Metabolome 2018 20180827) currently contains distributions of metabolite concentrations identified by NMR, and distributions of peak intensities of metabolites characterised by LC-MS detected in samples from an initial 10,719 volunteers (only 3012 for LC-MS so far).
For those interested in how to use quality controls in metabolomics the reader is directed to an article in Metabolomics that won this year’s prize for the most downloads—a testament that many researchers are aware that quality assurance and quality control is a very important aspect of any large-scale omics studies.9
Conclusions
So, for what the TMM project authors claim to be one of the biggest planned multi-omics longitudinal studies currently underway, it is clear that those with responsibility for the planning and execution of the project are certainly of the opinion that sampling really is critical to the quality of the whole omics project. Time will tell if they have taken enough precautions as the data sets increase in size and the analytical scientists start to use the resource to support the deployment of personalised medicine to these regions.
References
- http://www.rsc.org/events/detail/29574/challenges-in-analysis-of-complex-natural-mixtures-faraday-discussion (accessed 10 June 2019).
- G. Poste, “Bring on the biomarkers”, Nature 469, 156–157 (2011). https://doi.org/10.1038/469156a
- Y. Guo, A. Graber, R.N. McBurney and R. Balasubramanian, “Sample size and statistical power considerations in high-dimensionality data settings: a comparative study of classification algorithms”, BMC Bioinformatics 11, 447 (2010). https://doi.org/10.1186/1471-2105-11-447
- A. Kirpich, E.A. Ainsworth, J.M. Wedow, J.R.B. Newman, G. Michailidis and L.M. McIntyre, “Variable selection in omics data: A practical evaluation of small sample sizes”, PLoS ONE 13(6), e0197910 (2018). https://doi.org/10.1371/journal.pone.0197910
- D.K. Trivedi, K.A. Hollywood and R. Goodacre, “Metabolomics for the masses: the future of metabolomics in a personalized world”, Eur. J. Mol. Clin. Med. 3, 294–305 (2017). https://doi.org/10.1016/j.nhtm.2017.06.001
- C. Wu, F. Zhou, J. Ren, X. Li, Y. Jiang and S. Ma, “A selective review of multi-level omics data integration using variable selection”, High Throughput 8(1), 4 (2019). https://doi.org/10.3390/ht8010004
- D.I. Broadhurst and D.B. Kell, “Statistical strategies for avoiding false discoveries in metabolomics and related experiments”, Metabolomics 2, 171–196 (2006). https://doi.org/10.1007/s11306-006-0037-z
- S. Koshiba, I. Motoike, D. Saigusa, J. Inoue, M. Shirota, Y. Katoh, F. Katsuoka, I. Danjoh, A. Hozawa, S. Kuriyama, N. Minegishi, M. Nagasaki, T. Takai-Igarashi, S. Ogishima, N. Fuse, S. Kure, G. Tamiya, O. Tanabe, J. Yasuda, K. Kinoshita and M. Yamamoto, “Omics research project on prospective cohort studies from the Tohoku Medical Megabank Project”, Genes Cells 23(6), 406–417 (2018). https://doi.org/10.1111/gtc.12588
- D. Broadhurst, R. Goodacre, S.N. Reinke, J. Kuligowski, I.D. Wilson, M. Lewis and W.B. Dunn, ”Guidelines and considerations for the use of system suitability and quality control samples in mass spectrometry assays applied in untargeted clinical metabolomic studies”, Metabolomics 14, 72 (2018). https://doi.org/10.1007/s11306-018-1367-3