
 are removed via collision with anion in gas.")
A team of researchers lead by Kanazawa University have used a powerful molecular dynamics simulation to better understand the effect of excess charges on molecules tested by a mass spectrometer. They modelled the effect of adding molecules of the opposite charge in order to neutralise excess charge. In this case, the positive charge on polyethylene glycol (PEG) can be reduced via collision with negatively charged NO2– ions.
However, this is complicated by the fact that the likelihood of colliding depends on the amount of charge in the first place. “Charged polymers can adopt charge-state dependent structures because of electrostatic stretching”, first author Tomoya Tamadate says. For example, with small excess charge, PEG assumes a compact form. However, as the charge increased, the mutual repulsion between the positive charges causes it to straighten out.
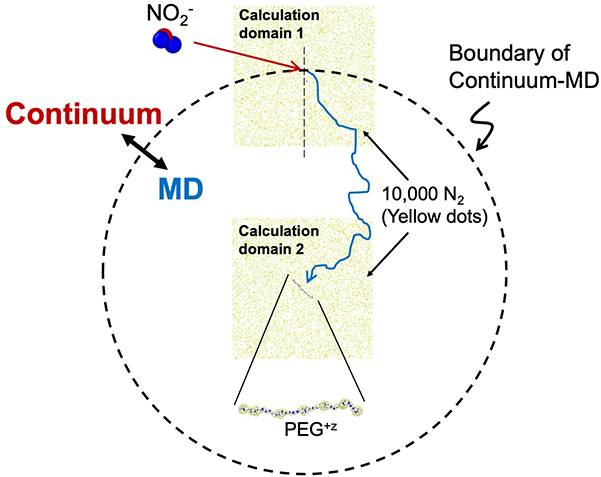
Figure 2. An outline of the developed calculation model (continuum-molecular dynamics simulation hybrid method). In this model, when the inter-ion distance is sufficiently large, relative motion is described by diffusion equations (continuum), meanwhile within a specific distance (shown as a broken line), molecular dynamics (MD) simulations are utilised to calculate the trajectory. In order to decrease the calculation cost, they perform the MD simulation with gas molecules only arranged around the target ions.
To help speed up the calculations, the team used the “continuum approximation” method, which only started simulating all of the atoms in the NO2– molecule once it approached close enough to the PEG.
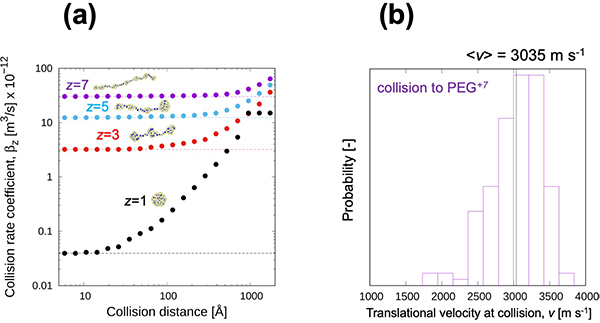
Figure 3. (a) The recombination (collision) rate coefficient of the PEG ion with different numbers of charges. This collision rate coefficient showed good agreement with the experimentally measured charge reduction rate. (b) Translational velocity distribution of NO2– ion at collision. We expect the kinetic energy can be used to evaluate the possibility of collision-induced reaction.
“The success of this project shows that hybrid continuum-molecular dynamics simulations can be used more generally to study collision-driven reactions molecules that can take on different conformations”, senior author Takafumi Seto says. The results can lead to more effective methods of controlling excess charge in sample molecules, which will allow for more accurate results.




, such as onions and garlic but also in the cruciferous family of vegetables (Brassicaceae), such as broccoli and cabbage. Credit: Osaka Metropolitan University")
 and Cryo-GC-FID (blue trace). Reproduced from https://doi.org/10.5194/amt-16-1061-2023 under a CC BY licence.")

