James A. Calladine and Michael W. George
School of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: [email protected]
Often, chemists are interested in the most efficient way of converting our starting materials into the desired product. A huge number of reactions proceed via the production of intermediate species, see Scheme 1, which are usually short-lived and difficult to detect. Such intermediates are considered to be of great importance because it is their reactivity which can determine the outcome of a reaction and, hence, the efficiency with which the final product is made. This can have widespread importance across the whole of chemistry, ranging from pharmaceutical and organic synthesis to catalysis and materials chemistry.

Many of the standard approaches to characterising compounds such as nuclear magnetic resonance (NMR) spectroscopy and mass spectrometry are not suited to probing very short-lived transient species in solution. However, a range of time-resolved techniques, such as luminescence, electronic and vibrational spectroscopies, can be effectively employed to observe and characterise these important and fleeting chemical intermediates.
There are two general approaches for studying short-lived chemical intermediates; (a) slowing the reaction down by lowering the temperature and using matrix isolation or inert solvents (such as liquefied noble gases) and characterising them using conventional spectroscopy, or (b) observing the reaction at room temperature occurring in “real-time” by fast time-resolved spectroscopy.1 Time-resolved techniques follow the course of a reaction by taking a snap-shot of the sample’s spectrum at various time intervals throughout the reaction. Many of the most fundamental chemical and biological transformations occur on very fast timescales and the ability to observe these is of great importance if we wish to understand how such basic processes happen. The results of matrix isolation, low temperature solution and time-resolved measurements are complementary, see Scheme 2, working together to provide a more complete mechanistic picture than either would individually.

Infrared (IR) spectroscopy is well suited to both observing and characterising transient intermediates in that it can provide crucial structural information. It is an ideal technique for following the reactions of metal carbonyl compounds, which can be important in catalysis. The ν(C=O) IR bands are excellent infrared chromophores with high extinction coefficients and their position is very sensitive to electron density at the metal centre. Here, we focus on fast time-resolved IR spectroscopic measurements and emphasise the implementation of an external cavity mid-IR quantum cascade laser (QCL),2 which provides easy-to-use apparatus for nanosecond time-resolved infrared spectroscopic measurements, in both glasses (77 K) and in solution (100–298 K).3
Many fast time-resolved IR spectroscopic measurements are based upon flash photolysis, a so-called “pump–probe” technique, whereby the intermediates of interest are formed by pumping the sample with a short UV or visible laser pulse and probing the outcome of this using IR spectroscopy. Time-resolved infrared spectroscopy (TR-IR) has a long history and, in 1958, Tanner and King reported rapid-scan IR measurements on the sub-millisecond timescale using a rapidly rotating Littrow mirror following photolysis of hydrazine in the gas phase.4 Since then, there have been many developments in this technique, as the approach can give information as to the fate of the parent starting material, the transient intermediates and any new products which may have been formed, along with their associated kinetics. Advances in TR-IR have often tracked developments in technology, particularly in lasers, detectors and data acquisition. Nowadays, ultrafast TR-IR spectra on picosecond or femtosecond timescales can be obtained using sophisticated femtosecond laser systems and difference frequency mixing to produce broadband IR pulses (ca 150–500 cm–1) which can be detected on multi-element mercury cadmium telluride (MCT) array detectors.5 The timing in such ultrafast IR laser systems is obtained using optical delay lines. Measurements can also be made on slower timescales by coupling the femtosecond IR laser to a high repetition rate nanosecond UV laser. The expensive nature of such laser systems can be prohibitive for utilisation in routine measurements.
The application of TR-IR spectroscopy to the study of condensed phase reaction mechanisms and transient intermediates followed the construction of an instrument by Friedrich Siebert and co-workers in Germany in the 1980s, which combined UV flash photolysis and a dispersive IR grating spectrometer with a fast risetime MCT IR detector. Spectra were built up on a “point-by-point” basis, with the change in IR transmittance following UV irradiation being measured at one particular frequency then repeated until the spectral range of interest was covered. By plotting the change in IR intensity against frequency for any required time-delay, a transient difference spectrum is created, see, for example, Figure 1.
 Example of TR-IR kinetic traces obtained using a lead salt IR diode laser source, showing the change in IR absorbance versus time at different IR frequencies (λ<sub>1</sub>, λ<sub>2</sub> and λ<sub>3</sub>) following the laser pulse (purple arrow). (b) TR-IR difference spectrum obtained at a particular time delay after the UV laser pulse. The spectral points obtained from the kinetic traces shown in (a) are highlighted in blue. (c) The incorporation of QCLs (left) into the Nottingham University point-by-point TR-IR step-up.")
These TR-IR spectrometers were based around a globar IR source, which suffers from the major problem of low IR photon flux. This can lead to a poor signal-to-noise ratio and resolution problems. Furthermore, atmospheric water absorbs strongly below ca 1950 cm–1 and even modest attenuation of the IR beam by the solvent used can cause a significant decrease in signal-to-noise. Nevertheless, developments by Hamaguchi and co-workers6 have shown that these problems can be overcome. By using a computer-controlled scanning dispersive spectrometer, coupled to an MCT detector with a low-noise preamplifier, they were able to detect very small transient signals (∆optical density, OD, ~ 10–6) on the nanosecond timescale across the mid-IR region. A more widely used approach for TR-IR spectroscopy on the nanosecond timescale is step-scan Fourier transform infrared (FT-IR) spectroscopy,7 which allows much easier data collection over wide frequency ranges. In step-scanning interferometry the mirror retardation is achieved in discrete steps, where the “moving” mirror is held still during data acquisition. Time-resolved data at each position in the interferogram can be recorded following an excitation event. The main disadvantage is that these measurements need to be highly repeatable as, typically, a large amount of averaging is needed to obtain a useable set of interferometric data.
Following Siebert’s original experiments, several groups started to use the point-by-point approach in the 1980s for TR-IR measurements.8 Early measurements were made using a continuous wave (CW) CO laser as the IR source. This laser is line-tunable in ca 4 cm–1 steps and can cover regions between ca 1550 cm–1 and 2100 cm–1, but the laser cavity must be cooled to provide the desired IR coverage and liquid N2 must be used in order to obtain lines up to 2100 cm–1. The main advantage of the CO laser over a globar is its much higher power (up to 1 W in a single laser line). However, a disadvantage in using this laser is its temperamental behaviour. This resulted in tunable lead salt (for example, Pb–Sn–Se) semiconductor IR diode lasers being the predominant IR source in TR-IR spectrometers. Although the IR output of these lasers is much lower than the CO laser (< 1 mW), they overcome the problem of spectral coverage since diodes are available throughout the mid-infrared region from 3 µm to 30 μm. However, these laser systems, as with those described above, still require cryogenic cooling (either 12–30 K or 77–100 K) and each individual laser has limited tunability (ca 100–150 cm–1).
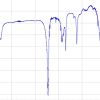
The most recent development in TR-IR has been the incorporation of a QCL into our point-by-point TR-IR system. This has enabled us to probe reactions with much greater sensitivity. The output power from a mid-IR external cavity QCL is much higher when compared to typical lead–salt diode lasers. Furthermore, operation at room temperature eliminates the need for cryogenic cooling, which is required for CO and IR diode lasers. The increased output power allows much higher signal-to-noise to be obtained and, in principle, enables the detection of species that are present at much lower concentrations. External cavity QCLs have broad spectral coverage and the output of the two lasers (Daylight Solutions Inc.) used at Nottingham University for TR-IR studies in the metal carbonyl region are shown in Figure 2. The use of these two QCLs provides continuous coverage over ca 250 cm–1.

We illustrate the use of these for the probing short-lived organometallic alkane complexes in solution both at room and low temperature.
Organometallic alkane complexes
The breaking (or activating) of C–H bonds in alkanes by transition metal complexes is of considerable interest since this offers an opportunity to utilise low cost feedstocks of hydrocarbons, such as natural gas, as a raw material for the chemical industry. It is becoming increasingly evident that, for many transition-metal mediated C–H activation reactions to take place, initial coordination of the alkane to the metal centre must occur, forming a so-called sigma–alkane complex. These are highly reactive and, as a result, tend to be very short-lived, but the nature of such organometallic alkane complexes can play a major role in determining the reaction outcome. These complexes were initially identified spectroscopically following irradiation of metal carbonyls in methane matrices at 12 K.9 Since then, time-resolved spectroscopy has been pivotal in characterising and determining their reactivity in solution.10
Of particular interest in this area are the organometallic complexes of rhenium. A breakthrough came following the report by Ball and co-workers of a long-lived Re–alkane complex, [CpRe(CO)2(alkane)], which was characterised by NMR spectroscopy.11 Since then, two other classes of Re–alkane complex have been identified through a combination of TR-IR and NMR spectroscopy. Other metal systems also show potential for this chemistry with Brookhart and co-workers recently reporting the first Rh–CH4 complex, [(2,6-(tBu2PO)2C5H3N)Rh(CH4)]+ to be characterised by NMR.12
We illustrate the use of fast TR-IR spectroscopy to detect organometallic alkane complexes by characterising the formation and reactivity of W-alkane complexes with a view to trying to characterise these using NMR spectroscopy, i.e. finding long-lived species that can be characterised by the much slower technique of NMR. Although there have been a large number of studies into Group VI metal carbonyls, the formation and reactivity of W(CO)5(cyclopentane) has not been reported. This is surprising, since it has been shown that cyclopentane forms some of the longest-lived organometallic alkane complexes. The TR-IR difference spectrum obtained 1 µs after photolysis of W(CO)6 in cyclopentane is shown in Figure 3(b).
 FT-IR spectrum of W(CO)<sub>6</sub> in cyclopentane solution, (b) TR-IR spectrum obtained 1 µs after the laser pulse (266 nm) and (c) typical single shot TR-IR kinetics showing the decay of W(CO)<sub>5</sub>(cyclopentane) at room temperature.")
The negative band shows that the parent ν(CO) IR band of W(CO)6 is bleached on irradiation and two new absorptions are produced at 1954 cm–1 and 1928 cm–1, which can readily be assigned to W(CO)5(cyclopentane). The bands of W(CO)5(cyclopentane) are formed within the 10 ns risetime of this TR-IR apparatus, which is limited solely by the MCT detector. W(CO)5(cyclopentane) is not stable and decays with a lifetime of a few hundred microseconds at room temperature. A typical single-shot TR-IR kinetic trace using the QCL source showing the decay of W(CO)5(cyclopentane) is presented in Figure 3(c). The reaction of W(CO)5(cyclopentane) (k2 = 8.9 × 105 M–1 s–1) shows this to be ca ×2 longer-lived than the corresponding n-heptane complex.
We have also measured the decay of W(CO)5(cyclopentane) in cyclopentane at 180 K by rapid-scan FT-IR. At 180 K, W(CO)5(cyclopentane) decays at a rate of kobs = 5.6 s–1, Figure 4(a). The lifetime of this alkane complex is, in principle, detectable using NMR spectroscopy; although this is currently at the detection limit of the technique.
We have also examined the reaction of an even shorter-lived first row Cr–cyclopentane complex, (η6–C6H6)Cr(CO)2(cyclopentane). The decay of the alkane complex is very fast (kobs = 50 s–1) and though spectral evidence for this can be provided by rapid-scan FT-IR, it was too short-lived to obtain reliable kinetics. Therefore, to obtain an accurate value of the lifetime of the alkane complex, the decay was measured using the QCL laser. Figure 4(b) shows the decay of the Cr–alkane complex at 180 K, recorded using the QCL and compares this to the decay of W(CO)5(cyclopentane) under similar conditions [Figure 4(a)]. As can be seen for lifetimes in the order of a few 10s of milliseconds, one can use single point QCL laser-based measurements to obtain excellent kinetic information when compared to the rapid-scan FT-IR, which can only record a few points in this short time interval.
{rokbox title=|Figure 4. (a) Decay of W(CO)5(cyclopentane) at 180 K recorded using rapid-scan FT-IR and (b) the decay of (η6–C6H6)Cr(CO)2(cyclopentane) at 180 K using the QCL.|}images/stories/articles/IR-21_6-F4.jpg{/rokbox}
Time-resolved infrared spectroscopy is a very useful technique for studying the reactions of short-lived intermediates. It can be used to give information on the structure and kinetic behaviour of a wide range of species, over a vast range of timescales, from femtoseconds to milliseconds. The development of this technique has relied on advances in technology. The use of QCL lasers offers an ideal IR source for nanosecond time-resolved infrared spectroscopy allowing sensitive TR-IR measurements, since they offer several advantages over other CW IR sources including high output power, good tunability and turn-key room temperature operation. TR-IR will hopefully be used more routinely and it is likely that QCLs will be useful for this purpose.
References
- R.A. Moss, M.S. Platz and M. Jones, Jr (Eds), Reactive Intermediate Chemistry. John Wiley & Sons, Chichester, UK (2004).
- E. Takeuchi, K. Thomas and T. Day, Laser Focus World 45(1), 83–86 (2009).
- D.C. Grills and M.W. George, in Handbook of Vibrational Spectroscopy, Vol. 1, Ed by J.M. Chalmers and P.R. Griffiths. John Wiley & Sons, Chichester, UK, pp. 677–693 (2002).
- K.N. Tanner and R.L. King, Nature 181, 963–965 (1958).
- M.D. Fayer (Ed.), Ultrafast Infrared and Raman Spectroscopy (Practical Spectroscopy). CRC Press, Cleveland, Ohio, USA (2001).
- T. Yuzawa, C. Kato, M.W. George and H. Hamaguchi, Appl. Spectrosc. 48, 684–686 (1994).
- G.D. Smith and R.A. Palmer, in Handbook of Vibrational Spectroscopy, Vol. 1, Ed by J.M. Chalmers and P.R. Griffiths. John Wiley & Sons, Chichester, UK, pp. 625–640 (2002).
- M. Poliakoff and E. Weitz, Acc. Chem. Res. 20, 408–414 (1987).
- R.N. Perutz and J.J. Turner, Inorg. Chem. 14, 262–270 (1975).
- A.J. Cowan and M.W. George, Coord. Chem. Rev. 252, 2504–2511 (2008) and references therein.
- S. Geftakis and G.E. Ball, J. Am. Chem. Soc. 120, 9953–9954 (1998).
- W.H. Bernskoetter, C.K. Schauer, K.I. Goldberg and M. Brookhart, Science 326, 553 (2009).