Rakesh Kandaa,* and Richard Glendinningb
aSevern Trent Services, Analytical Services, Britten Road, Reading, RG2 OAU, UK. E-mail: [email protected]
bSevern Trent Services, Analytical Services, Torrington Avenue, Coventry, CV4 9GU, UK
Introduction
The presence of chemical contaminants in the aquatic environment is of significant concern to the water industry due to possible health risks to humans, wildlife and domesticated animals. The source of chemical contamination of surface waters includes waste from industrial and domestic wastewater treatment plants, runoff from agricultural land and leachates from landfills and storage lagoons.
For countries within the European Union (EU), a number of directives provide a framework for environmental legislation which seeks to protect the environment and set water quality standards. Key directives include the Urban Waste Water Treatment Directive (UWWTD) which requires most towns and cities to treat their wastewater to specified standards and the Water Framework Directive (WFD). The WFD is the most substantial piece of water legislation ever produced by the EU; the directive requires all inland and coastal waters within defined river basin districts to reach good ecological and chemical status by 2015.1
Environmental regulations for the protection of the environment in the UK require monitoring of wastewater and environmental samples for a wide range of contaminants often at very low concentrations. For some compounds which have high biological activity, monitoring is required in the low parts per billion (µg L–1) to parts per trillion (ng L–1) range. Analysis of contaminants in wastewater at these low concentrations is difficult due to the complex composition of wastewater. Therefore, analytical techniques used to determine trace contaminants in wastewater need to be specific and allow analytes to be detected without interference from other compounds present in the sample (often at concentrations considerably higher than the compounds of interest). These requirements present a considerable challenge for traditional methods and, therefore, new procedures utilising advanced techniques such as high-resolution or tandem mass spectrometry have become common in environmental testing laboratories.
This article reviews the use of mass spectrometry (MS) in environmental and wastewater analysis.
Mass spectrometry
Mass spectrometry coupled to gas chromatography (GC) and liquid chromatography (LC) are ideal techniques for the determination of organic contaminants in wastewater due to the separation capability of GC and LC coupled to the selectivity and sensitivity of MS. This coupling of chromatography with mass selective detection allows the routine determination of many contaminants in parts-per-trillion (ppt) or lower concentrations. In recent years, developments in chromatography such as the introduction of ultra high-performance liquid chromatography (UPLC or UHPLC), which utilises the same separation methodology as conventional HPLC, but uses columns packed with smaller particles (generally about 2 µm particle size or less) has further enhanced the applicability of LC-MS in wastewater analysis.
The introduction of hybrid mass analysers such as the triple-stage quadrupole (TSQ) mass spectrometers allowed the use of tandem mass spectrometry (MS/MS) for quantitative low level analysis of analytes in complex mixtures or in the presence of a high sample matrix background. In the TSQ mass spectrometer, the first quadrupole (Q1) is connected with a collision cell (Q2) to another quadrupole (Q3). Both quadrupoles (Q1 and Q3) can be used in scanning or static mode, depending on the type of MS/MS analysis being performed. Types of analysis that can be performed include product ion scans, precursor ion scans neutral loss scan and selected reaction monitoring (SRM) also referred to as multiple reaction monitoring (MRM).
In MRM mode, Q1 is in static mode (precursor ion selected) and Q3 is also in static mode (product ion selected for the chosen precursor ion). The pair of ions selected is called a “transition” and multiple transitions form the basis for MRM in which different MRMs can be compared with analytical standards to provide quantitative and confirmatory analysis in a single analytical run. MRM is highly specific and virtually eliminates matrix background resulting in very selective analysis.
Gas chromatography mass spectrometry
GC was first coupled to MS at Dow Chemical Company with the aim of expanding the analytical capabilities of MS to cover complex mixtures of compounds.2 GC-MS has since become the technique of choice for volatile and semi-volatile, non-polar compounds including polycyclic aromatic hydrocarbons (PAH), polychlorinated biphenyls (PCB), pesticides, for example, organochlorine insecticides, dioxins, and other volatile priority pollutants. Compounds containing polar functional groups are difficult to analyse by GC, either because they are not sufficiently volatile, tail badly even on polar analytical columns, are thermally unstable and decompose in the GC or MS interface source.
Chemical derivatisation of samples prior to analysis can be used to overcome some of these problems. Derivatisation allows polar compounds to be modified to produce new compounds that have properties which are suitable for analysis using a GC-MS. Common chemical derivatisation methods include:
- silylation: used to volatilise compounds
- alkylation: used as the first step to further derivatisation or as a method of protection of certain active hydrogens
- acylation: used to add fluorinated groups
GC-MS is most often used in positive ion electron ionisation mode in either full scan (useful in determining unknown compounds in a sample or confirming compounds present) or in selected ion monitoring (SIM) mode in which only certain ion fragments specific to the analytes are monitored, allowing more scans to take place each second. Ion fragmentation patterns in electron ionisation (EI) are dependent upon the electron energy applied, typically 70 eV is used to facilitate comparison of generated spectra with library spectra.
Chemical ionisation (CI) is sometimes used for compounds chemically similar to those analysed by EI to enhance the abundance of the molecular ion resulting in less fragmentation. In positive ion CI a chemical reagent gas (for example, methane, ammonia, isobutene) is used. The reagent gas (R) is present at a much higher concentration than the analyte and is ionised by EI to give primary R+· reagent ions. The collision of the R+· ions with neutral R molecules lead to the formation of stable secondary ions, which are the reactant species that then ionise analyte molecules (A) by ion–molecule reactions.
Negative chemical ionisation (NCI), can be performed when an analyte contains electron-capturing moieties (for example, halogenated compounds). In negative ion CI, there are two primary mechanisms whereby negative ions are produced: electron capture and reactant ion chemical ionisation. Under CI conditions, electronegative molecules can capture electrons to generate negative ions. True negative ion chemical ionisation occurs by reaction of an analyte compound with negatively charged reactant ions (R–· or R–). Several types of ion–molecule reactions can occur, the most common being proton abstraction of the analyte resulting in the formation of (M – H)–. NCI can result in a significant increase in sensitivity (100 to 1000) over EI for certain electronegative compounds.
Liquid chromatography mass spectrometry
LC is a fundamental analytical technique in environmental analysis used for the determination of a wide range of compounds, including large molecular weight compounds and thermally labile compounds not suitable for GC. LC-MS is a rapidly growing technique for routine analysis of organic compounds in environmental analysis. For most compounds, a mass spectrometer is more sensitive and has greater specifity than other LC detectors.
Many of the advancements in LC-MS over the past decade have been in the development of ionisation techniques (interface/ion source). Earlier LC-MS systems used interfaces that either did not separate the mobile phase from analyte molecules (direct liquid inlet, thermospray) or did so before ionisation (particle beam). The analyte molecules were then ionised in the mass spectrometer under vacuum, often by traditional electron ionisation. These approaches had limited success for certain types of compounds.
The introduction of atmospheric pressure ionisation (API) techniques greatly expanded the applicability of LC-MS. In API LC-MS, the analyte molecules are ionised at atmospheric pressure. The analyte ions are then mechanically and electrostatically separated from neutral molecules.
Atmospheric pressure ionisation techniques are:
- electrospray ionisation (ESI)
- atmospheric pressure chemical ionisation (APCI)
- atmospheric pressure photoionisation (APPI)
ESI is used for the analysis of a wide range of compounds including large biomolecules. APCI is used for molecules less than 1500 u and is less well-suited than electrospray for analysis of large biomolecules and thermally unstable compounds. APCI is used with normal-phase chromatography more often than electrospray because the analytes are usually non-polar. APPI is applicable to many of the compounds that are suitable for analysis by APCI. APPI is particularly suitable for the analysis of highly nonpolar compounds. However, the most effective API technique for a particular application is not always easy to predict.
Environmental contaminants
There are various sources of environmental contaminants which include diffuse and point-source pollution originating from residential, commercial and industrial sources covering a wide range of chemicals and substances, including:
- nutrients
- heavy metals
- human pharmaceuticals
- veterinary medicines
- personal care products
- persistent organic pollutants, for example, pesticides, PCBs, PAHs
- flame retardants, for example, PFOS (perfluoroctane sulfonic acid), PFOA (perfluorooctanoic acid), PBDEs (polybrominated diphenyl ethers)
- nanoparticles
The WFD aims to establish a legal framework for the protection of water quality in European countries and recognises that specific measures have to be adopted regarding specific priority pollutants presenting a significant risk to the aquatic environment. The 33 substances and chemical compounds listed in Table 1 are included in the list of priority substances established by the EU. Some of these priority substances are also priority hazardous substances.
The WFD states that the emissions of priority substances need to be reduced progressively. The priority substances include 13 priority hazardous substances and the emission and discharge of these substances need to be removed or phased out completely.
The following substances and chemical compounds are required to be reviewed by the EU to determine if they should be included in the list of priority substances or priority hazardous substances: AMPA (aminomethylphosphonic acid); bentazon; bisphenol-A; dicofol; EDTA (ethylenediaminetetraacetic acid); free cyanide; glyphosate; mecoprop; musk xylene; PFOS; quinoxyfen; dioxins and PCB.
Analytical methodologies used to determine some environmental contaminants are described below.
Table 1. Water Framework Directive priority substances.
Number | Priority Substance | Identified as priority hazardous substance |
1 | Alachlor |
|
2 | Anthracene | Y |
3 | Atrazine |
|
4 | Benzene |
|
5 | Brominated diphenylether | Y |
6 | Cadmium and its compounds | Y |
7 | Chloroalkanes, C10-13 iv | Y |
8 | Chlorfenvinphos |
|
9 | Chlorpyrifos |
|
10 | 1,2-Dichloroethane |
|
11 | Dichloromethane |
|
12 | Di(2-ethylhexyl)phthalate (DEHP) |
|
13 | Diuron |
|
14 | Endosulfan | Y |
15 | Fluoranthenevi |
|
16 | Hexachlorobenzene | Y |
17 | Hexachlorobutadiene | Y |
18 | Hexachlorocyclohexane | Y |
19 | Isoproturon |
|
20 | Lead and its compounds |
|
21 | Mercury and its compounds | Y |
22 | Naphthalene |
|
23 | Nickel and its compounds |
|
24 | Nonylphenols | Y |
(4-nonylphenol) | Y | |
25 | Octylphenols |
|
26 | Pentachlorobenzene | Y |
27 | Pentachlorophenol |
|
28 | Polyaromatic hydrocarbons | Y Y Y Y Y Y |
29 | Simazine |
|
30 | Tributyltin compounds | Y |
(Tributyltin-cation) | Y | |
31 | Trichlorobenzenes |
|
32 | Trichloromethane (chloroform) |
|
33 | Trifluralin |
|
Endocrine disrupting compounds
The presence of trace quantities of endocrine disrupting chemicals (EDC) in the aquatic environment has become an issue of considerable concern. EDCs encompass a large range of compounds, many of which have been detected in wastewater and in the aquatic environment. EDCs have different types of activity (for example, estrogenic, androgenic and anti-androgenic) and cause their effects through different mechanisms of action, i.e. as agonists or antagonists or affect hormone synthesis and/or metabolism.3
EDCs can be divided into two categories: natural compounds, for example, hormones found in the bodies of humans and animals; phytoestrogen, which are substances found in some plants and man-made or industrial substances, which include a wide range of compounds—for example compounds that have been manufactured specifically to have endocrine effects, such as ethynyloestradiol used in the oral contraceptive pill. Some endocrine disrupting compounds found in the environment are breakdown products of other manufactured chemicals.
Research has shown that the natural oestrogens, oestrone (E1) and 17-b-oestradiol (E2) and the synthetic oestrogen 17-a-ethynyloestradiol (EE2) are responsible for most of the oestrogenic activity found in sewage effluents. The Environment Agency of England and Wales has proposed predicted no effect concentration (PNEC) values for all three free steroids expressed as annual averages of 0.1 ng L–1 (EE2), 1 ng L–1 (E2) and 3 ng L–1 (E1). Therefore, in order to test for compliance to these targets, very sensitive analytical methods are needed.
Typically, wastewater samples require extraction, concentration, clean-up and analysis using advanced analytical instrumentation. Samples are usually derivatised to produce non-polar derivatives prior to GC-MS/MS analysis but, when utilising LC-MS, the free steroids can be analysed directly after sample extraction and clean-up without the need for derivatisation. Various LC-MS techniques have been successfully applied to the analysis of steroid oestrogens including LC-QQQ, LC-ToF and LC-Orbitrap MS.
A method to determine steroid oestrogens in crude sewage, final treated effluent and surface waters has recently been applied in the UK for a large-scale monitoring programme.4 Water samples preserved with copper (II) nitrate/hydrochloric acid fixative were filtered to remove particulate matter prior to the addition of deuterated internal standards (2, 4, 16, 16-d4-estrone, 2, 4, 16, 16-d4-17-b-estradiol and 2, 4, 16, 16-d4-17-a-ethinylestradiol) and the sample was extracted onto a conditioned styrene divinyl benzene cartridge. The cartridge was dried under vacuum and then eluted with dichloromethane. The extract was then concentrated to 100 µL using blow-down equipment. The sample extract was cleaned up using gel permeation chromatography prior to analysis by LC-MS/MS. LC-MS/MS analysis was carried out using an Agilent 1100 LC and an AB Sciex API5000 mass spectrometer using negative ion electrospray ionisation in MRM mode. Positive identification of steroid estrogens was carried out by comparison of retention time and ion abundance ratios of the quantitation and confirmation mass transitions to that of a reference standard compound. Concentrations of steroid estrogens were calculated from a five-point calibration curve using internal standard quantitation. The limit of detection (LD) was 0.05 ng L–1 for all three estrogens.
Polybrominated diphenyl ethers
PBDEs are used as flame retardants in foam padding, plastics, fabrics, computer plastics, upholstered furniture, textiles, televisions and other products. PBDEs have been found to be persistent in the environment; they are bioaccumulative and have endocrine disrupting properties, particularly lower brominated molecules (Br 1-5), which can affect hormone levels in the thyroid gland. The commercial mixture of pentabromodiphenyl ether predominantly contains the penta-BDE congeners (50–62%), however, the mixture also contains tetra-BDEs (24–38%) and hexa-BDEs (4–8%), as well as traces of the tri-BDE (0–1%).
The EU Environmental Quality Standards (EQSs) for PBDEs is 0.5 ng L–1 (sum of all isomers). For reliable measurement of PBDEs at EQS levels, sensitive analytical methods are required which have a limit of quantification (LOQ) equal to or lower than 30% of EQS.
Standard methods for the determination of PBDEs in wastewater, soil and sludge are mostly based on solvent extraction and GC-MS analysis using electron ionisation or negative ion chemical ionisation. GC separation has to be carried out in such a way that sufficient separation of all PBDE congeners is achieved. Different gas chromatographic stationary phases could be applied for BDE determinations, but attention must be paid to potential interferences and co-elutions with other PBDE congeners, as well as with other brominated flame retardants, such as hexabromocyclododecane (HBCD), dimethyltetrabromobisphenol A (MeTBBPA) and polybrominated biphenyls (PBB), which are also present in many environmental samples.5
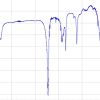
 Analysis of a mixture PBDEs using GC-MS/MS. (b) Sensitivity of PBDE 47 equivalent to 0.5 ng L–1 in an analytical standard (MRM transition 485.6 > 325.8).")
In a recent survey to determine PBDEs in treated wastewater samples as part of the UK’s Chemicals Investigation Programme, methods were developed to determine PBDEs with limits of detection of <0.1 ng L–1 using GC-MS/MS. Wastewater samples were analysed following addition of acetonitrile to the water sample to aid dissolution of the more insoluble PBDEs, followed by the addition of a mixture of internal standards. The samples were extracted using liquid–liquid extraction with n-hexane. Following extraction, a portion of the extract was concentrated using nitrogen blow-down and analysed by GC-MS/MS using selected MRM transitions with electron ionisation. Figure 1(a) shows the GC-MS separation of PBDE congeners using a J&W DB5MS 30 m × 0.25 mm × 0.1 µm fused silica capillary column. Figure 1(b) demonstrates the excellent sensitivity of GC-MS/MS for the analysis of a representative PBDE congener (PBDE 47) showing a signal-to-noise ratio of 103 at a concentration equivalent to 0.5 ng L–1 in a water sample.
- GC, combined with inductively coupled plasma mass spectrometry (ICP-MS), has also been successfully applied to the analysis of PBDEs. ICP-MS possesses several advantages:
- high sensitivity to PBDE congeners
- absolute selectivity between halogens, suffering no interference from fluorine, chlorine or iodine containing compounds, for example, PCBs
- no suppression of response from co-eluting compounds
- compound independent calibration (CIC), useful for the quantification of congeners for which standards are expensive or unavailable
Tributyltin
Organotin compounds are man-made compounds containing tin. The most used organotin compounds to date have been tributyltin (TBT) compounds which are substances containing the tributyltin moiety in their chemical structure. TBT uses include: wood preservation, as antifouling pesticides in marine paints (to protect from algal and barnacle growth), antifungal compounds in textiles and industrial water systems, such as cooling tower and refrigeration water systems. Tributyltin oxide is the most widely used compound in TBT-containing commercial products.
TBT is included in the UK Surface Waters (Dangerous Substances Classification) Regulations and the EU WFD Priority list substances. Organotin compounds can have adverse effects on marine organisms even at sub parts-per-trillion (ng L–1) levels, which is reflected in the very low environmental quality standard for TBT and its compounds of 0.2 ng L–1. These low limits are a challenge, even for the most sensitive analytical techniques such as GC-MS or GC-ICP-MS, therefore an extraction and pre-concentration step is invariably involved in their analysis.
Organotin compounds with less than four alkyl groups, for example, TBT compounds, are also too polar to analyse directly by GC and must be derivatised to form non-polar alkyltin compounds prior to analysis. In the past, most methods were based on extraction with troplone (a complexing agent) and n-hexane followed by derivatisation using Grignards reagent. Most standard methods in current use utilise in situ derivatisation of samples using tetraethylborate to produce ethyl organotin derivatives. The ethylated derivatives are extracted into hexane. A portion of the extract is then concentrated under nitrogen and analysed by large volume injection GC-MS. Figure 2 shows a mass chromatogram displaying the detection of TBT at 1.2 ng L–1 in a treated wastewater effluent sample in the presence of the labelled internal standard (d27-TBT).

Cypermethrin
Cypermethrin is a synthetic pyrethroid used as an insecticide in agricultural applications (in particular sheep dips) as well as in consumer products for domestic purposes. Freshwater EQS values for cypermethrin are 0.2 ng L–1 (long term) and 2 ng L–1 (short term). Sheep dip chemicals containing either cypermethrin or diazinon caused over a third of all environmental quality standard failures in England and Wales in freshwaters in 2006.
The low EQS for cypermethrin requires sensitive analytical methods. Most published methods to date utilise GC-ECD or GC-MS analysis. An issue with the analyses of cypermethrin at such low concentrations is that cypermethrin formulations consist of four geometric isomers, each of which can occur in two enantiomeric forms, hence eight possible peaks. With techniques utilising GC, usually only four peaks are detected for cypermethrin because the enantiomers (optical isomers) are not separated, therefore a detection limit lower than 50 pg L–1 is required for each peak.
Analytical methods to determine cypermethrin at these low levels therefore require large sample volumes (as high as 1 litre) to be extracted (by means of solid phase extraction or liquid–liquid extraction) or large volume injection is used with a smaller sample extraction volume. Alternatively GC-MS/MS in MRM mode can be used. GC-MS/MS provides greater selectivity and sensitivity than GC-MS.
Perfluorinated organic compounds
Fluorinated organic compounds, such as PFOS and related compounds including perfluorooctanoic acid (PFOA) and perfluorooctane sulfonamide (PFOSA) belong to a class of compounds known as perfluoroalkyl substances (PFAS).
PFOS and related substances are contained in cleaning products, fire fighting foams, carpets, textiles, paper and packaging, coating additives, leather and photographic materials. These substances have been shown to be persistent and bioaccumulative in the environment. They are highly toxic to certain species of wildlife, for example, honey bees. In the UK, the Environment Agency of England and Wales has undertaken a risk assessment and have given a proposed no effect concentration for freshwater of 25 µg L–1.
A method for the determination of PFOS and PFOA in wastewater samples has been developed.6,7 Samples are spiked with labelled internal standards and extracted using OASIS HLB (Waters Corporation) solid phase extraction cartridges and analysed using LC-MS/MS using negative ion electrospray in MRM mode, allowing limits of detection in the low ng L–1 to be achieved. Using multiple MRM transitions (Figure 3) for each analyte provides higher certainty of detection. Labelled internal standards are used to aid quantification, correcting for extraction recovery and ion suppression in wastewater samples.
 PFOS MRM transitions (498.8 > 80.0; 498.8 > 99.0) and 13C4-PFOS (502.8 > 99.0). (b): PFOA MRM transitions (413.0 > 369.0; 413.8 > 169.1 and 413.0 > 218.9 ) and 13C4-PFOA 417.0 > 169.1).")
Conclusion
New environmental legislation and the introduction of stringent environmental quality standards relating to environmental contaminants continues to provide significant analytical challenges to traditional methods of analysis. Advancements in mass spectrometry and associated chromatography instrumentation have become indispensible both in the research and routine analytical laboratory. Without some of the recent advancements in mass spectrometry many of the applications reviewed here would have not been possible just a decade ago without significant additional effort.
Recent advances in MS and availability of advanced analytical instrumentation have allowed analytical chemists to detect a vast range of substances in environmental and wastewater samples. Many of the advances are related to the introduction of high-resolution chromatography instrumentation, coupled to high resolution or tandem mass spectrometry instrumentation, improvements to ionisation sources, greater sensitivity of mass analysers and better acquisition software.
Advanced MS techniques combined with innovative sample preparation methods enable analytical chemists to have the ability to analyse and report substances at much lower concentrations than ever done previously in samples of high matrix complexity. Further advance in MS including the development and availability of techniques such as high-resolution time-of-flight and orbitrap MS instrumentation will continue to place mass spectrometry in the forefront of environmental analysis.
References
- European Union, Official Journal of the European Communities L327, 1–72 (2002).
- R.S. Gohlke and F.W.J. McLafferty, “Early gas chromatography/mass spectrometry”, J. Am. Soc. Mass Spectrom. 4, 367 (1993). doi: 10.1016/1044-0305(93)85001-E
- J.P. Sumpter, “Endocrine disrupters in the aquatic environment: an overview”, Acta Hydrochim. Hydrobiol. 33(1), 9–16 (2005). doi: 10.1002/aheh.200400555
- R. Kanda and J. Churchley, “Removal of endocrine disrupting compounds during conventional wastewater treatment”, Environ. Technol. 29(3), 315–323 (2008). doi: 10.1080/09593330802099874
- E. Eljarrat, A. de la Cal and D. Barceló, “Potential chlorinated and brominated interferences on the polybrominated diphenyl ether determinations by gas chromatography–mass spectrometry”, J. Chromatogr. A 1008, 181–192 (2003). doi: 10.1016/S0021-9673(03)00980-4
- N. Yamashita K. Kannan, S. Tanyiasu, Y. Horii, T. Okazawa, G. Petrick and T. Gamon, “Analysis of perfluorinated acids at parts-per-quadrillion levels in seawater using liquid chromatography-tandem mass spectrometry”, Environ. Sci. Technol. 38, 5522–5528 (2004). doi: 10.1021/es0492541
- C.L. McLaughlin, S. Blake, T. Hall, M. Harman, R. Kanda, J. Foster and P.C. Rumsby, Water Environ. J. 25, 13–21 (2011). doi: 10.1111/j.1747-6593.2009.00183.x