Elina Kalenius and Pirjo Vainiotalo
Department of Chemistry, University of Joensuu, PO Box 111, 80100 Joensuu, Finland. E-mail: [email protected]
During the last few decades, solution and solid state techniques have been utilised to obtain information about the properties of supramolecular host–guest complexes. Mass spectrometric analysis of these fragile non-covalent complexes has been focused on the determination of the molecular mass of the interacting molecules and the analysis has concentrated on the characterisation of covalent compounds. Since the invention of the soft ionisation techniques [namely ESI (electospray ionisation) and MALDI (matrix-assisted laser desorption/ionisation)] and their development for mass spectrometry (MS) instruments, the area and way that MS analysis is used have greatly changed and expanded. In particular, ESI1 has attained a steady position for the analysis of biomolecules, their non-covalent complexes and other rather fragile systems, which were earlier impossible to study by mass spectrometric methods.2,3 Today, MS can be employed not only for molecular weight identification purposes but also for sophisticated analyses on versatile properties of compounds.4 In the area of supramolecular chemistry, MS studies are becoming more and more general, although MS utilisation is still quite limited.
The main advantages of ESI-MS in the case of supramolecular complexes are its softness, sensitivity, specificity, speed and versatility. Fragile supramolecular complexes can undergo versatile analysis with low time and material consumption. Soft ionisation techniques allow even remarkably weak non-covalent complexes to be transferred to the gas phase from a variety of solvents as intact complexes with minimal sample consumption, and as a consequence their intrinsic properties can be studied without the interference of solvent. Properties such as stoichiometry of the self-assembly, relative binding affinities,5 the stability of the complexes and their gas-phase properties can be revealed.6 In fact, mass spectrometry can be used as a complete gas-phase “laboratory” covering the most important properties of non-covalent complexes in the absence of solvation interactions. Consequently, this also enables one to determine the importance of solvation interactions present in the solution. Moreover, MS analysis provides information about the gas-phase properties of the complexes, which is rather difficult or even impossible to obtain by other methods, in particular, for applications, in which supramolecular complexes are utilised in a gas-phase environment, this information is clearly vital.7
In this article we wish to highlight a few possibilities to exploit mass spectrometric analysis in supramolecular chemistry. We will give examples on competition experiments, dissociative experiments and ion–molecule reactions. Example spectra given here have been obtained by using a very sophisticated ESI-MS instrument, namely ESI-FT ICR MS (Fourier transform ion cyclotron resonance mass spectrometer).8 However, it must be emphasised that applications of these selected experiments also exist for other instrumental setups.
It has been stated that mass spectra reflect the situation in the solution state.9 Consequently, measurement of a single mass spectrum can, at best, reflect the affinities of the interacting counterparts and the thermodynamic equilibrium of the non-covalent complex formation. In Figure 1, ethanol complexation for the case of two different phosphonate cavitands is compared.10
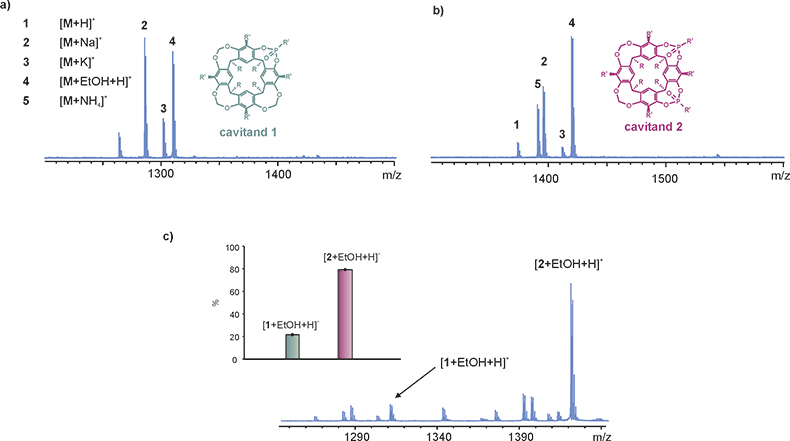
Figure 1. (a) Profile spectrum: cavitand 1 (2 µM) in TFA/EtOH/ACN (0.5/10/100), (b) profile spectrum: cavitand 2 (2 µM) in TFA/EtOH/ACN (0.5/10/100), and (c) right: competition spectra of cavitands 1 and 2 (2 µM, 1 : 1 in TFA/EtOH/ACN), left: averaged relative abundances of the competing complexes.10
There is a clear difference in the appearance of the spectra measured from solutions containing either monophosphonated cavitand 1 (Figure 1a) or diphosphonated cavitand 2 (Figure 1b). Both cavitands form a non-covalent complex [M + EtOH + H]+ in acidic acetonitrile solution containing ethanol. However, the ethanol complex formed with cavitand 2 clearly has a higher relative abundance as compared to cavitand 1. As shown in the competition experiment (Figure 1c), in which two cavitands are present in the same solution, the more abundant formation of the complex [2 + EtOH + H]+ is even more distinct. As mentioned, a simple spectrum can reflect the affinities of the interacting species in solution and experiments performed in a competitive environment can produce information about the affinities of the host compounds towards the selected guests. However, it must be emphasised that careful attention must be paid to the experimental arrangement and the conclusions deduced from the resulting spectra. There are several factors related mainly to the electrospray process which need to be taken into account. For reliable results, the competitive guests or hosts should be chemically and physically very similar and a set of analytes should be selected, where a particular property of the analytes can be strictly controlled. Differences in solvation, surface-activities, proton affinities, mass, unspecific complexation etc., can cause dramatic variation in the response of the complexes in electrospray ionisation and thereby results might be misleading.9,11 Moreover, compounds, such as additives and impurities present in solution, can alter the intensities of peaks as a consequence of a suppression effect or chemical interaction with analytes. In addition, it must be remembered that a mass spectrum never represents the universal truth about the characteristics of the sample solution—it only reflects characteristics of the ions present in the solution. However, as long as the experimental arrangement has been selected with great care, the competition experiments can reveal interesting information about the complex formation.
Today, there are several dissociative techniques available, which can be employed to study non-covalent supramolecular complexes: collision induced dissociation (CID), sustained off-resonance collision induced dissociation (SORI-CID), infrared multiphoton dissociation (IRMPD), electron capture dissociation (ECD) and electron transfer dissociation (ETD).12 The choice of which technique depends mainly on the available instrumentation and on the information sought. All these techniques are able to give structural information on the interacting molecules, e.g. information about the sub-structures the host or the guest molecule consists of. Additionally dissociative techniques can produce information on the relative kinetic stabilities of the complexes or possibly information on the site where the interaction takes place. Here we wish to highlight the information attainable by using CID, the most generally used dissociation technique. The CID experiment consists of four stages: 1, the ion of interest (predetermined m/z value) is selected and isolated from the profile spectrum; 2, the kinetic energy of the ion is increased by excitation; 3, the excited ion collides with inert background gas (i.e. Ar) and as a consequence a fraction of kinetic energy is transformed to internal energy, which induces the dissociation of the ion; 4, detection of the fragment ions. Depending on the instrumentation available, the kinetic energy increase can be controlled, which allows us to follow the dissociation of the ion as a function of energy. The dissociations of glucose and cellobiose complexes of tetraethyl resorcinarene are compared in Figure 2.13
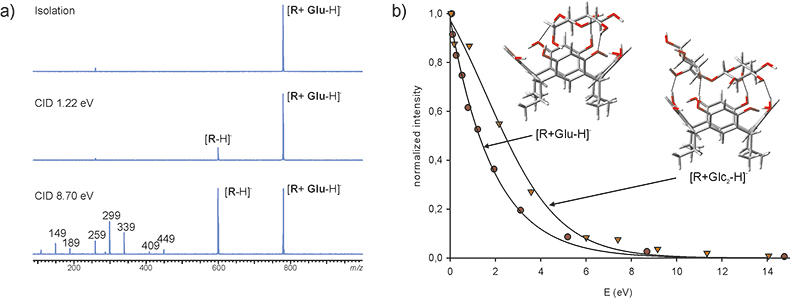
Figure 2. (a) CID of [R + Glu – H]– complex, and (b) comparison between the dissociation curves for [R + Glu – H]– and [R + Glc2 – H]– complexes.13
The resorcinarene forms easily deprotonated [M + saccharide – H]– ions with a variety of mono- and oligosaccharides. In the CID experiment, the increase in internal energy causes the dissociation of the non-covalent complex and the peak corresponding to [M – H]– appears in the spectrum (Figure 2a). As the energy is further increased the covalent bonds dissociate and also the fragment ions originating from the dissociation of the resorcinarene emerge. The dissociation of the complexes can be followed as a function of energy. The comparison between the decay of the peaks corresponding to the glucose and cellobiose complexes in an energy-resolved CID experiment is presented in Figure 2b. The dissociation curves reveal a clear stability difference between these two complexes. In that case, the stability difference originated from the number of hydrogen bonds formed in each complex, which was rationalised by using theoretical calculations. According to calculations the interaction energies (DE) for glucose and cellobiose complexes were 120 kJ mol–1 and 158 kJ mol–1, respectively. The difference between the interaction energies corresponds to the formation of one hydrogen bond more in the cellobiose complex as compared to the complex formed with glucose (Figure 2b).
Ion–molecule reactions have so far been only limitedly applied for studies of supramolecular complexes, although this type of experiment can produce significant and unique information about the reactivity, stability and conformations of supramolecular complexes in the gas phase.14 The ion–molecule reactions performed for supramolecular complexes can be divided into two categories: ligand-exchange reactions (guest-exchange) and H/D-exchange reactions. In ligand-exchange reactions an isolated complex ion reacts with a neutral reagent in the gas-phase and the ionic reaction products can be observed in the resulting spectrum. The stages of the experiment are: 1, isolation of the ion of interest; 2, its reaction with the neutral regent introduced to the reaction chamber (e.g. ICR cell or quadrupole); 3, the detection of the product ions. Ion–molecule reactions have been commonly performed using FT-ICR instruments, which allow the completion of long-time scale reactions. However, recently ion–molecule reactions have been increasingly performed by using other analysers, especially quadrupole ion traps (QIT), although in these instruments only relatively fast reactions can be studied.15 Depending on instrumentation the reactions can be followed as a function of time or as a function of the pressure of the neutral reagent. The selection of potential reagents is limited mainly by their volatility and reactivity.
Ligand-exchange reactions can produce information not only about the reactivity of the complexes, but also about the interactions involved, relative stability of the complexes and the steric factors related to complexation. Gas-phase H/D-exchange reactions are ion–molecule reactions performed using volatile deuterated reagents (commonly ND3, MeOD, D2O). As compared to H/D-exchange performed in solution, gas-phase H/D-exchange is more selective in reaction of labile hydrogens. It is expected that the H/D-exchange rate for hydrogen bonded or a shielded hydrogen is decreased or the reaction is even inhibited.16,17 Therefore, H/D-exchange in the gas-phase can be applied to study intra- or inter-molecular interactions of the labile hydrogens.
Figure 3 presents two examples from ion–molecule reactions of cavitand complexes with alkylammonium ions.17 In Figure 3a the ethylammonium complex of the monophosphonated cavitand reacts with neutral propylamine. In this reaction, a proton transfer occurs and as a product propylammonium complex of the cavitand is observed. Reactions were followed for 300 s. According to the acquired data, the reaction curves were drawn and the reaction rates were calculated. The cavitand complexes were also studied by using gas-phase H/D-exchange reactions (Figure 3b). Depending on the complex, all labile hydrogens were exchanged or the reaction did not occur.
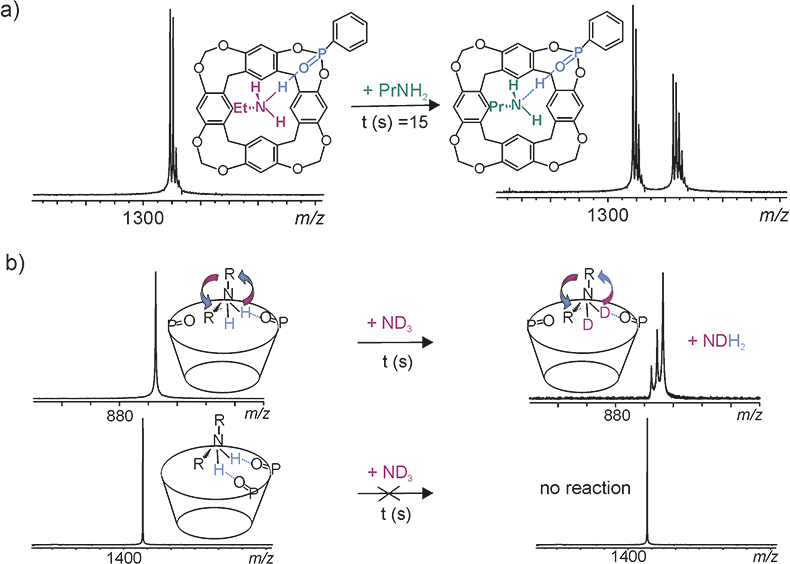
Figure 3. a) Ligand-exchange reaction with propylamine and b) H/D-exchange reaction with ND3.17
In both reaction types there were clear differences in reactions between different cavitand complexes. The ion–molecule reactions revealed that stabile, non-dynamic hydrogen bonding occurs only if the number of the available H-bonding donor sites equals the number of the acceptor sites. It was also discovered that the cavitands are able to utilise only two adjacent phosphonate groups simultaneously for interaction with alkyl ammonium ions. When this is not the case, the interaction resembles either a pendulum or a gyroscope, both compatible with the gas-phase ion–molecule reaction. The comparison gave vital information about the gas-phase reactivity and behaviour of these receptor systems, which are used in sensors operating on gas–solid interface.
References
- (a) M. Yamashita and J.B. Fenn, J. Phys. Chem. 88, 4451 (1984); (b) R. Cole (Ed.), Electrospray Ionization Mass Spectrometry. John Wiley & Sons, New York, USA (1997). https://doi.org/10.1021/j150664a002
- J.B. Fenn, M. Mann, C.K. Meng, S.F. Wong and C.G. Whitehouse, Science 246, 64 (1989). https://doi.org/10.1126/science.2675315
- M. Pryzybylski and M. Glocker, Angew. Chem. Int. Ed. Engl. 35, 806 (1996). https://doi.org/10.1002/anie.199608061
- C.A. Schalley (Ed.), Analytical Methods in Supramolecular Chemistry. Wiley-VCH, Weinheim (2007).
- J.S. Brodbelt, Int. J. Mass Spectrom. 200, 57 (2000). https://doi.org/10.1016/S1387-3806(00)00302-X
- J.S. Brodbelt and D.V. Dearden, Comprehensive Supramolecular Chemistry 8, 567 (1996).
- L. Pirondini and E. Dalcanale, Chem. Soc. Rev. 36, 695 (2007); (b) E. Dalcanale, M. Melegari, M. Suman, L. Pirondini, D. Moiani, C. Massera, F. Ugozzoli, E. Kalenius, P. Vainiotalo, J.-C. Mulatier and J.-P. Dutasta, Chem. Eur. J., in press.
- Bruker BioApex 47e FT-ICR, equipped with an InfinityTM cell, a 4.7 Tesla 160 mm bore superconducting magnet, and an external ApolloTM ESI source.
- C.A. Schalley, Int. J. Mass Spectrom. 194, 11 (2000). https://doi.org/10.1016/S1387-3806(99)00243-2
- E. Ventola, P. Vainiotalo, M. Suman and E. Dalcanale, J. Am. Soc. Mass Spectrom. 17, 213 (2006). https://doi.org/10.1016/j.jasms.2005.10.017
- J.M. Daniel, S.D. Friess, S. Rajagopalan, S. Wendt and R. Zenobi, Int. J. Mass Spectrom. 216, 1 (2002). https://doi.org/10.1016/S1387-3806(02)00585-7
- E. de Hoffmann and V. Stroobant, Mass Spectrometry, Principles and Applications. Wiley, Chichester, UK, pp. 190–201 (2007).
- E. Kalenius, T. Kekäläinen, R. Neitola, K. Beyeh, K. Rissanen and P. Vainiotalo, Chem. Eur. J. 14, 5220 (2008).
- D.V. Dearden, Y. Liang, J.B. Nicoll and K.A. Kellersberger, J. Mass Spectrom. 36, 989 (2001); (b) E. Ventola, K. Rissanen and P. Vainiotalo, Chem. Commun. 1110 (2002); (c) G. Grigorean, S. Gronert and C.B. Lebrilla, Int. J. Mass Spectrom. 219, 79 (2002); (d) E. Ventola, K. Rissanen and P. Vainiotalo, Chem. Eur. J. 10, 6152 (2004); (e) U. Mazurek, O. Geller, C. Lifshitz, M.A. McFarland, A.G. Marshall and B.G. Reuben, J. Phys. Chem. A 109, 2107 (2005).
- J.E. Chipuk and J.S. Brodbelt, Int. J. Mass Spectrom. 267, 98 (2007). https://doi.org/10.1016/j.ijms.2007.02.026
- E. Ventola, A. Hyyryläinen and P. Vainiotalo, Rapid Commun. Mass Spectrom. 20, 1218 (2006). https://doi.org/10.1002/rcm.2440
- E. Kalenius, D. Moiani, E. Dalcanale and P. Vainiotalo, Chem. Commun. 3865 (2007). https://doi.org/10.1039/b707842k