M. Mohamed, A.G. Cox and C.W. McLeod*
Centre for Analytical Sciences, Department of Chemistry, University of Sheffield, Sheffield S3 7HF, UK. E-mail: [email protected]
Introduction
There is considerable interest in chemical imaging of pharmaceutical tablets since knowledge of the spatial distribution of constituents is critical to ensuring uniformity and consistency of product. Pharmaceutical tablets, in general, are complex multicomponent blends comprising active ingredients(s) and a variety of inactive substances—the excipients—that are used to aid manufacture and facilitate tablet administration. Thus, in addition to measurement of the spatial distribution of the active drug, there is a need to monitor excipients such as binders, fillers, coatings, lubricants, disintegrants and preservatives. Imaging of organic and inorganic constituents of tablets represents a considerable challenge and no single spectroscopic approach can provide definitive characterisation of all components and/or satisfy key measurement criteria such as sensitivity, specificity, resolution and speed of analysis. With respect to molecular imaging, Fourier transform infrared (FT-IR), Raman and fluorescence microscopies are widely used in the pharmaceutical industry. Indeed efforts have been made to exploit the complementary nature of IR and Raman by merging respective data sets in order “to enable a more complete visualisation of pharmaceutical formulations”. More generally the approach of Clarke et al.1 termed “Chemical Imaging Fusion” can be extended to elemental imaging given that inorganic compounds and heteroatoms are critical components of formulations.
Progress in elemental imaging of pharmaceutical tablets has been restricted to a few specialised techniques. Scanning electron microscopy with energy-dispersive X-ray analysis (SEM/EDAX) attachment represents the state of the art for high resolution measurement, but the high capital cost, need for vacuum and extended analysis times limit its use to non-routine investigations. Commercially available instrumentation based on micro-X-ray fluorescence (µ-XRF)2 and laser-induced breakdown spectroscopy (LIBS)3 have been developed for tablet characterisation and imaging but, to date, there are only a few published reports. In the case of µ-XRF2 distribution maps were realised for a suite of elements—Na, Mg, Si, P, S, Br, Ca, Fe, Co, Zn, Bi—in tablets using an X-ray probe beam of 60 µm. Due to spectral complexity, multivariate statistical analysis was used to improve visualisation and interpretation of elemental correlations within samples. The authors concluded that both “elemental as well as molecular imaging techniques are needed to make a full characterisation of the distribution of all tablet constituents”.
In the LIBS technique a high energy laser beam (Nd:Yag, 1064 nm) is used to ablate and excite atomic/ionic energy states prior to measurement of time-resolved emission spectra (wavelength range 190–850 nm). Unlike µ-XRF the laser ablation (LA) process is destructive resulting in the production of small craters when the laser is fired continuously at a fixed site. Laser spot sizes of 50–500 µm are typical in a LIBS experiment. The scope for LIBS in tablet characterisation is well illustrated in the work of Bechard and Mouget,3 which includes detailed studies on blend and tablet uniformity, film coating uniformity, dose strength and drug/component mapping.
The laser ablation technique can be linked directly to inductively coupled plasma (ICP) excitation sources, i.e. ICP emission and ICP mass spectrometry (ICP-MS) and provides much improved performance over LIBS, particularly in respect of sensitivity and signal quantification. Attractive features of the ICP technique include wide elemental coverage, high sensitivity (sub ppb/ppt), high specificity, wide dynamic range and simultaneous/near simultaneous measurement capability. This together with the advantages of laser sampling, i.e. minimal or no sample preparation, and the ability to probe samples with laser beam diameters of a few microns to hundreds of microns ensures a powerful and versatile microanalytical facility. The LA-ICP technique has been applied to pharmaceutical tablet analysis although the possibility for imaging was not considered.4
The last few years has witnessed the development of laser ablation ICP-MS as a new elemental imaging modality. Much of this activity has been in the life sciences and most applications relate to imaging of biomedical tissue sections with respect to the distribution of trace and toxic elements,5,6 Gd-based magnetic resonance imaging (MRI) contrast agents7 and protein biomarkers labelled with elemental centres.8,9 Elemental imaging via LA-ICP-MS has also been applied to plant10 and metallurgical systems.11 Given the successful use of LA in diverse imaging applications there is clear scope for extending investigations to pharmaceutical tablets. In this work a direct-reading ICP emission spectrometer is used for elemental detection as an alternative to ICP-MS principally because the ICP emission technique is more robust than ICP-MS and because the enhanced sensitivity of ICP-MS is not required.
Instrumentation
The experimental system consisted of a Nd:Yag laser ablation system (New Wave ESI MACRO, 266 nm) interfaced to a direct-reading ICP atomic emission spectrometer (ICP-AES) (Spectro Analytical, ARCOS). As illustrated in Figure 1 the LA cell has provision for computer-controlled x-y-z translation, which allows for relatively large areas (1–4 cm2) of the sample to be interrogated through multiple line rastering. Ablated material, in the form of an aerosol, is swept into the ICP discharge by a stream of argon carrier gas and a direct reading emission spectrometer with high data acquisition rate (100 ms) is used for simultaneous time-resolved measurement of the atomic emission of elements present in the sample. In this way it is possible to acquire multielemental data as a function of spatial co-ordinates (x-y). Imaging software is then used to transform the data set into distribution maps for selected elements. Table 1 lists typical operating conditions for LA-ICP-AES measurement.

Table 1. Operating parameters for LA-ICP-AES.
| Laser ablation (New Wave Macro 266 nm) Laser energy: 2 mJ at 10 Hz Beam diameter: 80—240 µm Line raster rate: 60 µm s–1 |
| ICP-AES (Spectro Analytical ARCOS SOP) ICP Power: 1400 W Auxiliary Argon: 1 L min–1 Coolant Argon: 12 L min–1 Carrier Argon: 0.85 L min–1 Analytical lines (nm): Al (167.078), B (208.959), Br (148.845), C (193.091), Ca (183.801), Cl (134.724), Cu (224.700), Fe (238.204), I (178.276), Mg (279.079), Mn (257.611), Na (330.237), P (177.495), Si (251.612), Se (196.090), S (180.731), Ti (307.864), Zn (206.201) Signal integration time: 100 ms Imaging software: Graphis (Kylebank) |
Elemental imaging
Method development
ICP-AES is a powerful multielement analysis technique that was originally developed for liquid sample analysis. The use of laser ablation for sample introduction considerably extends analytical capabilities permitting direct analysis of solids at the sub ppm level. Given that a high percentage of marketed drugs contain heteroelements such as C, Cl, Br, F, S, Na and a wide range of inorganic additives as part of the formulation there is considerable scope for utilising LA-ICP-AES as a new imaging tool.3
To assess the scope for elemental imaging, studies were first directed at an over-the-counter nutritional supplement, Osteocare, which contains Ca and Mg and a range of trace elements. It was projected that the Osteocare tablet would serve as an internal control material. Specifically it was suspected that the material would exhibit inhomogeneous elemental distributions that would be detected through laser ablation interrogation. Prior to commencing laser ablation studies, bulk chemical analysis of Osteocare tablets was performed by conventional ICP analysis to establish chemical composition. As shown in Table 2 elements were present at widely different concentrations and, in general, determined values were consistent with the manufacturers’ specification.
Table 2. Multi-element analysis of Osteocare tablet (after acid dissolution)
| Element | Specified conc. (mg/tablet) | Measured values | ||
| mg/tablet | μg g–1 | Recovery % | ||
| B | 0.3 | 0.370 | 239 | 123 |
| Ca | 400 | 359 | 231759 | 90 |
| Cu | 0.5 | 0.543 | 350 | 108 |
| Fe | — | 0.298 | 192 | — |
| Mg | 150 | 147 | 94899 | 98 |
| Mn | 0.25 | 0.231 | 149 | 92 |
| Se | 0.025 | 0.025 | 16 | 100 |
| Zn | 5 | 4.8 | 3099 | 96 |
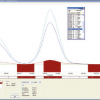
As part of method development, systematic studies were performed to identify analytically useful laser operating parameters. The approach adopted was to vary either laser beam diameter or laser energy and study the effect on elemental response; other laser operating parameters being held constant (see Table 1). Figure 2 shows the effect of variation in laser energy level on Ca emission (183.8 nm); each of the emission–time response curves corresponds to a single line raster across the tablet. It can be seen that there is a progressive increase in signal intensity as laser energy is increased, consistent with an increase in mass transfer to the ICP. It can also be seen that the laser generated signal is not constant suggesting a non-uniform Ca distribution.


Figure 3 presents the variation in signal response as a function of laser beam diameter, a laser energy level of 60% being used for the experiments. Emission–time responses for the four line raster scans were not smooth, again suggesting non-homogeneity of Ca. Analyte sensitivity was dependent on laser beam diameter, the improved sensitivity at wide beam diameter, e.g. 240 µm, reflecting an increase in ablated material. On the basis of the above results it is clear that there is a compromise between attainable sensitivity and spatial resolution, nevertheless, it is concluded that laser interrogation (line rastering mode) of pharmaceutical tablets provides a powerful route for detecting analyte heterogeneities. Subsequent work was directed at multielemental imaging based on multiple line rastering of samples.
 for Osteocare.")
Images for 12 elements—Mg, Ca, Fe, Si, Al, Mn, Zn, Na, Cu, S, B, Se—are presented in Figure 4. The images were based on multiple line rastering (n = 16) with a laser beam diameter of 240 µm and with a separation distance of 240 µm between adjacent line rasters, i.e. the non-interrogated zone width was 240 µm. Total analysis time was about 100 minutes.
Visual examination of the maps suggests wide differences in the degree of analyte homogeneity and with no element exhibiting a uniform distribution. In the case of Fe, Zn, Na, Cu, S, B and Se it would appear that these elements were present in the formulation as distinct particles or agglomerates of relatively large size (up to 1 mm). It is also possible to infer chemical associations for the elements by noting correlations in the variation of signal with spatial location; thus Na is associated with B, Zn with S and Al with Si, respectively. This is further apparent from examination of the respective emission–time responses (single line rasters) for the elements Zn and S.
A quantitative measure of the degree of analyte heterogeneity was obtained by calculating measurement precision [% RSD (% relative standard deviation)] for the variation of analyte emission intensities for the duration of a single line raster. The precision data of Table 3 range from about 9% RSD (Al) to 174% RSD (Cu). Given that measurement precision for analysis of a homogeneous material by LA-ICP-AES would be of the order 2–5% RSD it can be concluded that Osteocare or similar material could serve as a control sample for benchmarking analyte homogeneity in candidate formulations. A further point to emphasise regarding the analytical capability of LA-ICP-AES for elemental imaging is that the technique is equally effective at monitoring trace (Se) and major level components (Ca).
Table 3. Measurement precision for single line rastering (Osteocare tablet).
| Element | Al | B | Ca | Cu | Fe | Mg | Mn | Na | S | Se | Si | Zn |
| % RSD | 9 | 148 | 21 | 174 | 11 | 19 | 53 | 84 | 89 | 63 | 10 | 161 |
Survey analysis
To test the general applicability of LA-ICP-AES for imaging of pharmaceutical tablets, measurements were extended to a small selection of prescription medicines. Figure 5 presents results for Maferol, an Fe-based supplement used in the treatment of anaemia. Visual examination of the images suggests a relatively homogeneous distribution for the active component (Fe) and the elements monitored—Al, C, Fe, Mg, Mn, Si. In general, the emission–time responses for the elements were essentially steady-state as represented in the single-line rasters (Figure 5) which contrasts sharply with previous data for Osterocare. Measurement precision (RSD) was in the range 3–26%.


The heteroatom I was used to monitor the anti-arythmic drug amiodarone (C25H29I2NO3; RMM 645.3). As shown in Figure 6, the image and respective line raster for I indicate a homogeneous drug distribution. The results for C atomic emission are also consistent with a uniform drug distribution but caution must be exercised in using this element as a diagnostic, since organic compounds are also present in formulations as excipients. Magnesium stearate is a key excipient in formulations and homogeneity was checked via measurement of Mg. Visual examination of the image indicates a non-uniform distribution in comparison to that for the drug. Measurement precisions for single line rasters were: Mg 18%, I 8% and C 4%.
In a final example, elemental composition and homogeneity was assessed for tablets produced by two different manufacturers. The active substance in each, Sotalol (C12H20N2O3S; RMM 272.4), contains a S heteroatom which permits spatially resolved measurement of the drug. Multielement analysis of the tablets by conventional ICP analysis (i.e. after acid dissolution) was first performed and results (analytical data not shown) indicated major differences in formulation chemistry. The elemental images are presented in Figure 7 and it is clear that the drug is relatively homogeneous in both products as indicated by the uniformity in S response (%RSDs for single line raster: Sotacor, 8%; Sotalol, 5%). A major difference in chemical composition relates to Ca and P where these elements are present in Sotalol but absent in Sotacor. Moreover, there is a strong correlation in emission signals for Ca and P which suggests that calcium phosphate is used as a tablet binder in Sotalol. In contrast to the results for S, measurement precision for Ca and P was poor (%RSDs: Ca 56% and P 58%) signifying a non-homogeneous distribution for calcium phosphate.

Conclusion
Laser ablation in combination with ICP emission spectrometry represents a powerful new tool for imaging elemental distribution in pharmaceutical tablets. The approach, applicable to both inorganic and organic constituents (provided there is a measurable elemental centre) affords high sensitivity, high specificity, wide elemental coverage and good spatial resolution. Moreover, the quantitative nature of ICP spectrometry ensures a robust measure of analyte homogeneity/heterogeneity through calculation of %RSD values. Elemental imaging via laser rastering is relatively time consuming, however, many analytical situations that require tablet characterisation would suffice with single line rastering or point ablation. Although not addressed in this study LA-ICP-AES is applicable also to depth profiling and impurity analysis.
Acknowledgement
The authors are grateful to Dr D. Talib for technical drawing.
References
- F.C. Clarke, M.J. Jamieson, D.A. Clark, S.V. Hammond, R.D. Jee and A.C. Moffat, Anal. Chem. 73, 2213–2220 (2001). https://doi.org/10.1021/ac001327l
- T.C. Miller and G.J. Havrilla, Powder Diffr. 20, 153–157 (2005). https://doi.org/10.1154/1.1913720
- S. Bechard and Y. Mouget, “LIBS for analysis of pharmaceutical materials”, in Laser Induced Breakdown Spectroscopy—Fundamentals and Applications, Ed by A.W. Miziolek, V. Palleschi and I. Schechter. Cambridge University Press (2006).
- R. Lam and E. Salin, J. Anal. At. Spectrom. 19, 938–940 (2004). https://doi.org/10.1039/b314732k
- J.S. Becker, M. Zoriy, B. Wu, A. Matusch and J. Su Becker, J. Anal. At. Spectrom. 23, 1275–1280 (2008). https://doi.org/10.1039/b805228j
- J.S. Becker, M. Zoriy, A. Matusch, D. Salber, C. Palm and J. Su Becker, Mass Spectrom. Rev. 29, 156–175 (2010).
- N. Kamaly, J.A. Pugh, T.L. Kalber, J. Bunch, A.D. Miller, C.W. McLeod and J.D. Bell, Mol. Imaging Biol. https://doi.org/10.1007/s11307-009-0282-4
- R. Hutchinson, A.G. Cox, C.W. McLeod, P.S. Marshall, A. Harper, E.L. Dawson and D.R. Howlett, Anal. Biochem. 346, 225–233 (2005). https://doi.org/10.1016/j.ab.2005.08.024
- J. Seuma, J. Bunch, A.G. Cox, C.W. McLeod, J. Bell and C. Murray, Proteomics 8, 3775–3784 (2008). https://doi.org/10.1002/pmic.200800167
- J.S. Becker, R.C. Dietrich, A. Matusch, D. Pozebon and V.L. Dressler, Spectrochim. Acta 63B, 1248–1252 (2008). https://doi.org/10.1016/j.sab.2008.08.001
- C. Dubuisson, A.G. Cox, C.W. McLeod, P. Krause, I. Whiteside, R. Jowitt and H. Falk, Iron and Steel Institute of Japan International 44, 1868–1875 (2004). https://doi.org/10.2355/isijinternational.44.1859