Markus Furger and Suzanne Visser
Laboratory of Atmospheric Chemistry, Paul Scherrer Institute, 5232 Villigen PSI, Switzerland. E-mail: [email protected]
Introduction
Ambient aerosols consist of a wide variety of elements and compounds that can either condense to form clusters and particles up to sizes of tens of micrometres, or that were already abraded from a surface by some mechanical process. These particles remain suspended within the atmosphere for a time that depends on their particle size, weight, solubility and other physical factors. While airborne, they exert an influence on the Earth’s radiation budget and, hence, the Earth’s climate, or they may be inhaled and deposited within the respiratory tract of animals or human beings, where they may have an adverse effect on health.1 Health effects depend on particle size and chemical composition; larger particles penetrate less deeply into the lung and are more easily removed from the respiratory tract. The connection between particle size and human health has been demonstrated in many studies, but the effect of chemical composition on health is less well known and still remains a subject of study. Apart from aromatic compounds that are carcinogenic, various elements have adverse health effects caused by their toxicity. Such elements are typically found in aerosols, in only trace concentrations and, hence, they are not easy to measure. Nevertheless, a deeper understanding of the concentrations and spatial and temporal distribution of trace elements is required to identify their emission sources and to develop strategies for their reduction and mitigation. In this article, we will discuss the determination of trace element concentrations in ambient aerosols by synchrotron radiation-induced X-ray fluorescence spectrometry.
Instrumentation
We collected ambient aerosols by means of three-stage rotating drum impactors (RDIs). Air is sucked through the different stages, which are separated by nozzles, with a pre-set flow rate of 1 m3 h–1 that determines the particle size cut-off for each stage. The pre-selector at the air inlet prevents particles larger than 10 µm from entering the instrument. The cut-off size at each drum is determined by the flow speed and the nozzle dimension. The RDI deposits by impacting particles with diameters between 2.5 µm and 10 µm on the first, 1.0–2.5 µm on the second and 0.1–1.0 µm on the third stage.2,3 We will denote the first, second and third stages as coarse, intermediate and fine size fractions, respectively. Other instruments, with more stages and different cut-off characteristics, are available in other research groups. The selected particles are deposited on Apiezon-L-coated 6.5 µm polypropylene (PP) foil mounted on aluminium drums. The drums are held fixed in position during the sampling interval and then they are rotated by 3.5° to the next sampling position. This generates a barcode-like pattern on the foil. Sampling intervals are, typically, one to two hours. With 2 h sampling, one set of drums lasts for eight days (or 3 × 96 samples) after which they have to be replaced with empty ones. The used drums are then stored in plastic containers at room temperature until they are analysed. Our RDIs have been deployed in recent years in field campaigns in Zurich (Switzerland),4,5,6 Barcelona (Spain),7 Paris (France), Pasadena (USA), Marseille (France) and London (UK).
Analytical approach
From typical European cities, the 2 h sampling yields deposits on the PP foil of the order of 1 µg of total mass (distributed over the three stages). Much of the collected aerosols are sulfates, nitrates, organic species and black carbon. Trace elements merely constitute a few percent of the total aerosol mass, which means they often amount to only a few nanograms. These low concentrations and the large number of samples collected in a typical field campaign require an analytical technique that is sensitive enough to measure low concentrations while at the same time does not demand excessive sample treatment. Therefore, synchrotron radiation-induced X-ray fluorescence (SR-XRF) spectrometry has become our method of choice.8 Sample spectra can be obtained within 30 s of X-ray exposure.
We measure the elements from sodium to zinc in vacuum with monochromatic light at the Swiss Light Source (SLS) in Villigen, Switzerland and the elements from titanium to lead in air with a white beam at HASYLAB/DESY in Hamburg, Germany (Figure 1, Table 1).

Figure 1. (a) Beamline set-up at the SLS, showing the vacuum chamber with the RDI sample holder inside (not visible) and the silicon drift X-ray detector (towards the left of the image) at a 90° angle relative to the incident beam (which enters from the top-right corner). (b) Beamline set-up at HASYLAB, showing the RDI sample holder on the rotating table in the centre, the Si(Li) detector in the background. The beam enters from the right.
Table 1. Beamline characteristics at the Swiss Light Source (SLS) and at HASYLAB/DESY for X-ray fluorescence measurements.
| SLS | HASYLAB/DESY |
Beamline | X05DA (Optics) | L |
Energy | 10.5 keV, monochromatic | 45 keV, white beam |
Analysed elements | Na (Z = 11) to Zn (Z = 30) | Ti (Z = 22) to Pb (Z = 82) |
Detector | SDD (Si) | Si(Li) |
Atmosphere | Vacuum | Air |
Typical beam spot size | 70 × 140 µm | 80 × 150 µm |
Mass calibration is done with mixtures of element standards printed with an inkjet printer on the same PP foil as used for sampling, a technique inspired by a research group in Hamburg.9 For the absolute calibration, the printed foil is analysed with inductively coupled plasma optical emission spectrometry (ICP-OES) and the resulting mass per analysed area is related to the signal obtained by SR-XRF.
The measured spectra are analysed with the open source software PyMca10 and with Canberra’s WinAxil Version 4.00 program.
Results and discussion
To illustrate the advantages of SR-XRF, data from the MEGAPOLI project (Megacities: Emissions, urban, regional and Global Atmospheric POLlution and climate effects and Integrated tools for assessment and mitigation),11 are presented here. The winter field campaign took place from 15 January to 15 February 2010 in the greater Paris, France, area. Ambient air was sampled simultaneously with RDIs deployed at two sites, one (LHVP) near the city centre, the other at the SIRTA site in Palaiseau, about 20 km south-west of the first site and in an urban background setting. Figure 2(a) shows the time series from the SIRTA site of sodium in the three size ranges. For most elements, including sodium, most of the mass is deposited in the coarse size fraction and least is deposited in the fine fraction. This is true for most of the time, but an exception is observed on day 26.6, where the intermediate fraction exceeds the coarse fraction, while the fine fraction is equal to the coarse fraction. Comparable values were observed at the city site [Figure 2(d)], except for the coarse fraction which is missing due to bad handling of the sample holder during a synchrotron beamtime use. The sodium mass deposition in the intermediate fraction on that day is related to the zinc time series, as shown in the lowest part of Figure 2(c) and indicates a different source than for the rest of the campaign. The absence of a chlorine peak at that time further corroborates this. Sodium is an excellent tracer for maritime air masses which transport sea salt over the continent, or for de-icing salt applied to roads in wintertime. Both sources produce particles in the coarse and the intermediate size fractions, but practically none in the fine fraction. The good agreement between the two sites [Figure 2(d)] for most of the campaign gives a preference towards sea salt as the main (regional) source. The situation on day 26 is characterised by easterly winds and a massive drop in temperature and pressure (not shown), but no precipitation. The prominent fine fraction of zinc hints to industrial or combustion processes. It can be hypothesised that this peak in fine zinc concentrations is the result of the change in air mass and advection of industrial emissions from the east across the city. The 2 h time resolution of the RDI samples does not clearly indicate the difference in arrival times of the plume at the LHVP site and at the SIRTA site, which was estimated from the observed wind speed to be roughly an hour.
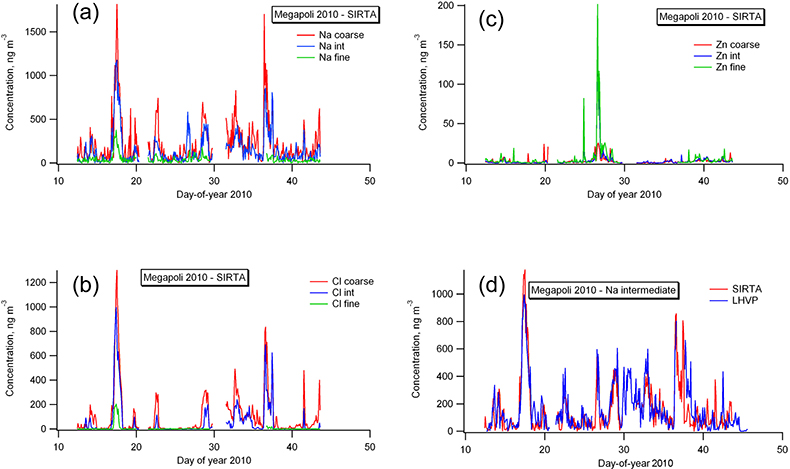
Figure 2. MEGAPOLI 2010 winter campaign concentration time series in ng m–3 of (a) sodium (Na), (b) chlorine (Cl) and (c) zinc (Zn) in three size ranges (coarse, intermediate, fine; see text for details) for the SIRTA site about 20 km from the city centre of Paris. (d) Comparison of the intermediate size fraction of sodium between the LHVP and SIRTA sites.
At this time, we can only speculate about the real source of the zinc peak on day 26. The data presented here are in a preliminary state of analysis. A more detailed source apportionment includes all available elements and auxiliary data from other co-located instruments, as well as backward trajectory analyses from meteorological fields. These data are analysed with statistical methods (for example, see Reference 12). Such analyses will be the next steps performed with this dataset. The results will extend the basis for the understanding of the complex processes in atmospheric chemistry in a megacity.
Concluding remarks
Synchrotron radiation-induced X-ray fluorescence spectrometry is a very capable tool with which to analyse minuscule amounts of matter collected with high time resolution. It is extremely sensitive and by far outweighs conventional methods with respect to analysis time and sample preparation. A caveat, however, is the restricted access to synchrotron facilities which requires success in the application for allocation of beamtime.
Acknowledgements
The research leading to these results has received funding from the Swiss National Science Foundation, grant 200021_132467, from FP/2007-2011 under grant agreement 212520 and from the European Community’s Seventh Framework Program (FP7/2007-2013) under grant agreement no. 312284.
References
- F.J. Kelly and J.C. Fussell, “Size, source and chemical composition as determinants of toxicity attributable to ambient particulate matter”, Atmos. Environ. 60, 504 (2012). doi: 10.1016/j.atmosenv.2012.06.039
- N. Bukowiecki, A. Richard, M. Furger, E. Weingartner, M. Aguirre, T. Huthwelker, P. Lienemann, R. Gehrig and U. Baltensperger, “Deposition uniformity and particle size distribution of ambient aerosol collected with a rotating drum impactor”, Aerosol Sci. Technol. 43, 891 (2009). doi: 10.1080/02786820903002431
- A. Richard, N. Bukowiecki, P. Lienemann, M. Furger, M. Fierz, M.C. Minguillón, B. Weideli, R. Figi, U. Flechsig, K. Appel, A.S.H. Prévôt and U. Baltensperger, “Quantitative sampling and analysis of trace elements in atmospheric aerosols: impactor characterization and synchrotron-XRF mass calibration”, Atmos. Meas. Tech. 3(5), 1473 (2010). doi: 10.5194/amt-3-1473-2010
- N. Bukowiecki, P. Lienemann, M. Hill, R. Figi, A. Richard, M. Furger, K. Rickers, G. Falkenberg, Y. Zhao, S.S. Cliff, A.S.H. Prevot, U. Baltensperger, B. Buchmann and R. Gehrig, “Real-world emission factors for antimony and other brake wear related trace elements: Size-segregated values for light and heavy duty vehicles”, Environ. Sci. Technol. 43(21), 8072 (2009). doi: 10.1021/es9006096
- N. Bukowiecki, P. Lienemann, M. Hill, M. Furger, A. Richard, F. Amato, A.S.H. Prévôt, U. Baltensperger, B. Buchmann and R. Gehrig, “PM10 emission factors for non-exhaust particles generated by road traffic in an urban street canyon and along a freeway in Switzerland”, Atmos. Environ. 44(19), 2330 (2010). doi: 10.1016/j.atmosenv.2010.03.039
- A. Richard, M.F.D. Gianini, C. Mohr, M. Furger, N. Bukowiecki, M.C. Minguillón, P. Lienemann, U. Flechsig, K. Appel, P.F. DeCarlo, M.F. Heringa, R. Chirico, U. Baltensperger and A.S.H. Prévôt, “Source apportionment of size and time resolved trace elements and organic aerosols from an urban courtyard site in Switzerland”, Atmos. Chem. Phys. 11(17), 8945 (2011). doi: 10.5194/acp-11-8945-2011
- T. Moreno, X. Querol, A. Alastuey, C. Reche, M. Cusack, F. Amato, M. Pandolfi, J. Pey, A. Richard, A.S.H. Prévôt, M. Furger and W. Gibbons, “Variations in time and space of trace metal aerosol concentrations in urban areas and their surroundings”, Atmos. Chem. Phys. 11(17), 9415 (2011). doi: 10.5194/acp-11-9415-2011
- N. Bukowiecki, P. Lienemann, C.N. Zwicky, M. Furger, A. Richard, G. Falkenberg, K. Rickers, D. Grolimund, C. Borca, M. Hill, R. Gehrig and U. Baltensperger, “X-ray fluorescence spectrometry for high throughput analysis of atmospheric aerosol samples: The benefits of synchrotron X-rays”, Spectrochim. Acta B 63, 929 (2008). doi: 10.1016/j.sab.2008.05.006
- U.E.A. Fittschen and G.J. Havrilla, “Picoliter droplet deposition using a prototype picoliter pipette: Control parameters and application in micro X-ray fluorescence”, Anal. Chem. 82(1), 297 (2010). doi: 10.1021/ac901979p
- V.A. Solé, E. Papillon, M. Cotte, P. Walter and J. Susini, “A multiplatform code for the analysis of energy-dispersive X-ray fluorescence spectra”, Spectrochim. Acta Part B 62(1), 63 (2007). doi: 10.1016/j.sab.2006.12.002
- F. Freutel, J. Schneider, F. Drewnick, S.L. von der Weiden-Reinmüller, M. Crippa, A.S.H. Prévôt, U. Baltensperger, L. Poulain, A. Wiedensohler, J. Sciare, R. Sarda-Estève, J.F. Burkhart, S. Eckhardt, A. Stohl, V. Gros, A. Colomb, V. Michoud, J.F. Doussin, A. Borbon, M. Haeffelin, Y. Morille, M. Beekmann and S. Borrmann, “Aerosol particle measurements at three stationary sites in the megacity of Paris during summer 2009: meteorology and air mass origin dominate aerosol particle composition and size distribution”, Atmos. Chem. Phys. 13(2), 933 (2013). doi: 10.5194/acp-13-933-2013
- V.A. Lanz, A.S.H. Prévôt, M.R. Alfarra, S. Weimer, C. Mohr, P.F. DeCarlo, M.F.D. Gianini, C. Hueglin, J. Schneider, O. Favez, B. D’Anna, C. George and U. Baltensperger, “Characterization of aerosol chemical composition with aerosol mass spectrometry in Central Europe: an overview”, Atmos. Chem. Phys. 10(21), 10453 (2010). doi: 10.5194/acp-10-10453-2010