A.N. Davies
External Professor, University of Glamorgan, UK, Director, ALIS Ltd, and ALIS GmbH – Analytical Laboratory Informatics Solutions
“The man who gets the most satisfactory results is not always the man with the most brilliant single mind, but rather the man who can best coordinate the brains and talents of his associates.”—W. Alton Jones
Background
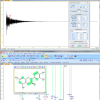
There has been much debate about which program can predict NMR spectra the best. It is well known within the NMR community that spectra prediction strongly depends on the “quality” of the starting data sets for those systems which use real data as a knowledge base. It has become a hot topic in some blogs, although disappointingly most of the authors tend to have affiliations to one software vendor or another.
Predictions on the whole need to be much more accurate in 1H NMR spectroscopy as opposed to 13C NMR in order to help the scientist. In general the prediction software available to date does a pretty good job when the results of the predictions are used sensibly by well-trained and experienced spectroscopists who recognise the pitfalls in this field and use the predictions only to help their general expert assessment of the results in front of them.
What has been surprising in the recent debate is the nature of the claims and counter-claims for “supremacy” when the absolute improvements in prediction “accuracy” are often less than the errors or normal variation expected between compounds measured on different instruments using different procedures, different solvents or concentrations.
As with most analytical techniques, the volume of reference data from which to draw hard and fast rules is negligible compared to the breadth of chemistry which a technique may have to span. Different strategies for 1H NMR spectra prediction have different strengths and weaknesses, but no particular approach dominates. The most common algorithms used for NMR prediction either draw on rules generated by studying the peer-reviewed literature values for chemical shifts or are reference database-based, where measured spectra and their assigned two-dimensional or three-dimensional chemical structures are the source knowledge base for training systems such as neural networks. I recently heard of an approach to 1H NMR prediction which seems to actually try for an obvious improvement.