
Nelson R. Alexander
Roche Diagnostic Solutions, Tucson, AZ, USA
DOI: https://doi.org/10.1255/sew.2021.a20
© 2021 The Author
Published under a Creative Commons BY-NC-ND licence
Editor’s introduction: There has been a long wait for the Theory of Sampling (TOS) to make any inroads within the medical profession. Indeed, a few intrepid explorers have tried, but this has resulted only in getting nowhere fast… so far. This column lays out what can only be described as a paradigm shift within the field of tumour diagnostics, nothing less. And it all came about because of a young scientist’s inspired application of the TOS; indeed “all that was needed” was… a blender. Here our readers are treated to a fascinating summary of what happened, and how. Talk about structured heterogeneity; talk about a clean break with grab sampling; talk about inspired use of blending—this column is a shear TOS delight!
A lightbulb moment
Thirty seconds after I was told by Kate Leith, PhD, to “get a blender” I realised she was right. By the time I returned to my desk I had stopped thinking the suggestion was the most ridiculous thing I had ever heard, and began wondering about where I was going to get the right blender. You see I am a cancer biologist looking to solve a specific spatial heterogeneity problem, and Dr Leith is a biostatistician who is an expert in statistical power analysis. In the days following the sentence that forever changed my career back in 2015, I came to better understand Pierre Gy’s Theory of Sampling (TOS) and have been working to apply representative sampling to cancer diagnostics ever since.
Background: cancer biology
A tumour develops from a single cell that has incurred enough DNA mutations for the cell to begin to proliferate uncontrollably, as the normal checks and balances within a cell are broken. From that single cell, a tumour composed of billions and billions of cancer cells begins to form. However, as the tumour grows, individual cancer cells continue to gain unique mutations. Some of these unique mutations will aid in tumour development and, therefore, that cancer cell will generate more cells. This process happens over and over within a growing tumour and can generate many distinct populations of cancer cells, with each population containing unique mutations. Thus, solid tumours are a heterogeneous mass of distinct cancer cell populations: a process cancer biologists call tumour heterogeneity.
A great analogy for thinking about tumour heterogeneity, borrowed from my collaborator Dr Charles Swanton, is a tree. The trunk of the tree represents all of the DNA mutations found in every cancer cell within a tumour because they were the first set of mutations that led to the formation of the tumour. The bottom branches represent early DNA mutations that generated distinct populations of cancer cells with unique growth properties within the tumour. As you move up the tree, each branch is yet another set of DNA mutations generating yet another distinct population of cancer cells within the tumour. The movement from the bottom of the tree to the top represents the lifespan of the tumour, such that the base of the tree is the initiating cell, and the leaves are the most recent cells. In this way, a palm tree is similar to a tumour with a very recent burst of DNA mutations (low tumour heterogeneity), whereas an oak tree represents a tumour with significant DNA mutations throughout the lifespan of tumour development (very high tumour heterogeneity).
How it is done traditionally
While readers of this column are very familiar with Gy’s TOS, and have applied the kind of composite sampling needed to counteract spatial heterogeneity many times over, we in cancer biology have only come to appreciate the full scale of this problem over the last decade. While many researchers had understood that cancer cells within a tumour are not identical for over a century, only after the technology that enabled the sequencing of the human genome was applied to tumours did we fully appreciate the scale of the heterogeneity of solid tumours. In fact, the initial paper that demonstrated the sampling problem in solid tumour diagnostics was published in 2012,1 amazingly recent given the long history of the practice of medicine.
The endeavour of cancer biology is well over 100 years old, and many of the techniques and methods used to sample and process tumours date back to the late 1800s. These innovations enabled scientists and doctors to cut out portions of a tumour, cut them into thin slices (less than half the diameter of a strand of hair), and place them on a glass slide for microscopic examination. This approach has served the medical profession, and patients well—but they are not fully optimised as we came to realise.
The same instruments and methods are still used today to provide samples for cancer diagnosis and more advanced complex DNA-based tests. Applying this sampling approach to the tumour tree analogy, the current solid tumour sampling method is akin to taking the trunk and a single branch from an oak tree. We can tell it is an oak tree, and can likely determine what DNA mutations caused the first branch to form, but we are blind to the rest of the tree. To continue the tree analogy, once we have taken the sample of the tree and determined what type of tree it is, we burn the rest. Quite literally, unused surgical tumour tissue is routinely and universally incinerated. My lab takes tumour tissue that would otherwise be discarded and uses it as the input for representative sampling through homogenisation.
What’s new
In collaboration with teams led by Dr Samra Turajlic at the Francis Crick Research Institute and the Royal Marsden Hospital, NHS Foundation Trust, we published the first manuscript describing our new representative sampling approach for solid tumours in 2020.2 In this article we compared homogenisation of tumour tissue to many individual standard biopsy samples taken from different spatial locations (65 in total), all from the same tumour. A standard tissue biopsy sample is akin to drilling out a core sample in soil or a drill core through a mineralised rock (ore)—a cylindrical excision from solid tissue.
In what may not be all that surprising to practitioners of the TOS, representative sampling of solid tumour tissue enables the detection of more DNA mutations than do individual samples. Representative sampling also generates reproducible aliquots of the original homogenised sample, whereas each individual standard biopsy sample will contain different combinations of DNA mutations (Figure 1). We are very excited about this new approach and are currently working to expand these findings to more tumours and across more tumour types (we originally focused on kidney cancer).
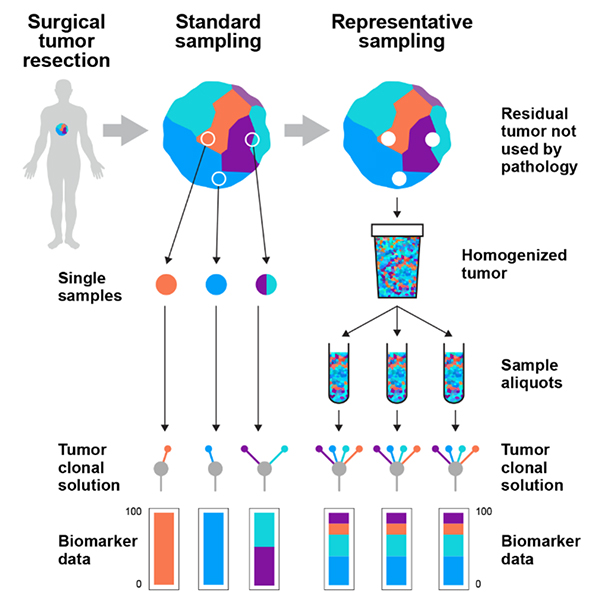
Figure 1. Tumours are spatially heterogeneous mixtures of multiple populations of cancer cells with distinct DNA mutations. Current sampling strategies fail to address the heterogeneity of tumours as individual biopsy samples cannot capture the diversity of the entire tumour—they are in fact just grab samples. These standard samples generate biased data depending on the area of the tumour that was sampled, being blind to the area that was not sampled. Representative sampling through blending of the residual tumour tissue not taken by pathologists creates a homogenate that contains the full diversity of the original solid tumour mass. Reproduced from K. Litchfield et al., “Representative sequencing: unbiased sampling of solid tumor tissue”, Cell Rep. 31(5), 107550 (2020).
Further refinement
Another aspect of the TOS that we have integrated into our work (although I must admit I only realised this after the fact) is defining the appropriate particle size for analysis of a heterogeneous lot.3 The tumour tissue that we are homogenising is transformed from a pliant soft material to a very ridged and dense material through a process called formalin fixation. Homogenising fixed tissue in a blender (very similar to, or possibly the same blender you have at home in your kitchen) generates fragments of tumour tissue containing hundreds to thousands of cells. We can take that homogenate and immediately purify DNA or protein molecules by taking a sub-sample of the homogenate and breaking down all of the molecules into a liquid, and specifically purifying just the DNA. However, as the unit of heterogeneity within our sample is a single cell, we wanted to be able to further process the homogenate into single cells.
Next, we developed a protocol to break the fragments of tumour from the homogenate into individual nuclei. The nucleus of a cell contains the DNA molecules that we are testing, so nuclei are an appropriate proxy for individual cells. We can then assess the characteristics of millions of nuclei taken from a representative sample of a tumour, and collect only the nuclei from cancer cells, separating them away from the nuclei of other normal cells that innervate the solid tumour mass. This process dramatically improves our analytic sensitivity to DNA mutations because we are testing predominantly cancer nuclei, rather than a combination of cancer and normal nuclei (which do not have the DNA mutations).
Impact on patient care: personalised healthcare
In solid tumours, every distinct population of cancer cells could harbour genetic mutations that confer resistance to therapeutics. Armed with this knowledge, over the past few decades, treatment strategies have evolved from treating all patients with the same type of cancer with the same drug, to linking specific DNA alterations to specific drugs. This strategy is called personalised healthcare and has produced amazing results for some patients. Researchers and clinicians continue to look for new personalised targets of therapy in hopes that the personalised healthcare strategy can expand to include more and more patients. Fundamental to this work is our ability to find mutations that can be linked to specific therapies, hence applying the TOS to heterogeneous tumours is critical.
My initial conversations with Kate (“pre-TOS enlightenment”) were simply thought experiments, focused on determining “how many individual samples were needed to detect DNA mutations present in a small portion of a large tumour”. At this point I was “functionally fixed” to the current standards, hoping that if we simply did more of what we currently do, we could solve the sampling problem.
The problem with this line of thought is that it misses one of the most important aspects of the TOS. In order to be able to design a sampling plan—“how much” should be sampled and “how”—one must first understand, and be able to characterise, the level of heterogeneity present in the original sample material. This intuitive feature of the TOS has had the most impact on my thoughts around how solid tumours should be sampled. There is no way of knowing upfront how heterogeneous a solid tumour is at the level of DNA. Yet linking the right treatment to the right patient requires that we detect as many of the DNA mutations within a tumour as possible.
Therefore, in solid tumour oncology, representative sampling is truly a matter of life or death.
References
- M. Gerlinger et al., “Intratumor heterogeneity and branched evolution revealed by multiregion sequencing”, New Engl. J. Med. 366, 883–892 (2012). https://doi.org/10.1056/NEJMoa1113205
- K. Litchfield et al., “Representative sequencing: unbiased sampling of solid tumor tissue”, Cell Rep. 31(5), 107550 (2020). https://doi.org/10.1016/j.celrep.2020.107550
- L. Petersen, P. Minkkinen and K.H. Esbensen, “Representative sampling for reliable data analysis: Theory of Sampling”, Chemometr. Intell. Lab. Syst. 77(1–2), 261–277 (2005). https://doi.org/10.1016/j.chemolab.2004.09.013

Nelson Alexander
Nelson Alexander received his PhD in Cancer Biology from the University of Arizona in 2005, followed by a post-doctoral fellowship at Vanderbilt University for two years. Wanting to have a more direct impact on patient care, he joined Roche Tissue Diagnostics (RTD) in 2007 where he has held multiple roles in R&D, as well as in Research & Early Development. During his time at RTD, Nelson became very interested in the interface between tumour heterogeneity and sampling bias. Nelson currently leads a research team within Roche Diagnostic Solutions that is pioneering a new sampling process that enables evaluation and quantification of tumour diversity at the genomic, transcriptomic and cellular levels from entire solid tumours.
[email protected]
