
John P. Hammond
Technical Manager, Starna Scientific Limited, 52–54 Fowler Road, Hainault, Essex IG6 3UT, UK
DOI: https://doi.org/10.1255/sew.2022.a4
© 2022 The Author
Published under a Creative Commons BY-NC-ND licence
Introduction
As stated in the last article, this article marks a change in this series because from this point forward, we will concentrate on specific areas of interest in the Quality environment in which we are/will be working in the future.
As an unexpected consequence of the COVID-19 Pandemic, the enforced isolation of the last two years has allowed time for reflection in all aspects of life. In the scientific area, the postponement or reassignment to virtual events of major international conferences, e.g. Pittcon 2022 at the time of writing this article, has prompted an explosion of on-line webinars, presentations etc. on a whole host of topics, and the Quality environment is no exception. Available to a much wider audience than by physical attendance at a conference, this “information explosion” in these areas of interest is summarised below and reinforced by the author’s direct involvement in these areas of interest.
Regular readers of this Quality Matters column will be familiar with the regulatory environments and standards often discussed by the authors; and in that respect this series of articles is no different. Discussed below are two new areas of interest, their associated terminology, standards and guidance on the topic.
The “new” quality “tools” of Analysis of Risk and Lifecycle Management
Whilst neither of these concepts can be described as new in the fundamental definition of the term, their use and application in our areas of interest certainly is.
Analysis of Risk
In a simple definition, risk may be defined as the combination of the probability of occurrence of harm and the severity of that harm. However, achieving a shared understanding of the application of risk management among diverse interested parties is difficult because each stakeholder might perceive different potential harms, place a different probability on each harm occurring and attribute different severities to each harm. In addition, subjectivity can directly impact the effectiveness of risk management activities and the decisions made.
However, as stated below, in regulated environments there is clearly an increasing perception as to the benefit of the analysis of risk associated with any given process, and, therefore, risk management principles are effectively utilised in many areas of business and government including finance, insurance, occupational safety, public health, pharmacovigilance and by agencies regulating these industries.
In recent years, certaintly since the publication of the revised ISO/IEC 17025:2017 standard,1 “Risk Management” is one of these new topics. It is included in ISO/IEC 17025 as section “8.5 - Actions to address risks and opportunities” where specifically in 8.5.2 it states:
8.5.2 The laboratory shall plan:
a) actions to address these risks and opportunities.
b) how to:
— integrate and implement these actions into its management system.
— evaluate the effectiveness of these actions.
NOTE Although this document specifies that the laboratory plans actions to address risks, there is no requirement for formal methods for risk management or a documented risk management process. Laboratories can decide whether or not to develop a more extensive risk management methodology than is required by this document, e.g., through the application of other guidance or standards.
So, we now have “consideration of risk” incorporated into this fundamental quality standard, and whilst the note states that there is no requirement for formal risk management, we live in an evolving World, and I’ll leave you to draw your own conclusions.
So, this has elevated the role of Risk Management in modern Quality Management systems, because just like we have learnt to distinguish between “Variants of Interest” and “Variants of Concern” in this pandemic, “guidance” is often perceived exactly as stated, i.e. additional information for consideration, but as soon as this becomes stated as a requirement within a standard, it takes on a whole new meaning.
On a related topic, it will be interesting to see how the ISO remit of ISO TC/334 to convert the ISO/REMCO Guides into corresponding standards is perceived, but that’s a discussion for another article/day.
Thereafter, two more recently published documents also for consideration are ISO 31000:20182 and ICH Q9(R1).3
To assist organisations in implementing risk management in a structured manner, ISO have added “ISO 31000:2018 – Risk management – A practical guide” to their portfolio of standards, and have made freely available in electronic format (.pdf) a guidance document for ISO 31000:2018.4 The aim of this standard is to assist organisations seeking guidance on how to integrate risk management into their activities. The handbook, therefore, includes information on risk management principles, the framework, roles and responsibilities, planning, processes, communication, monitoring and review, and continual improvement; and was written for those who are either starting their risk management journey or require additional guidance on how to improve their current, risk management programme.
In the standard itself, ISO 31000 explains the fundamental concepts and principles of risk management, describes a framework, and outlines the processes of risk identification and management. ISO 31000 is supplemented by IEC 31010:2019, “Risk management – Risk assessment techniques” and ISO 31073, “Risk management – Vocabulary”, and, therefore, with this set of three standards, effective risk management can be planned, and implemented, using the appropriate structures and terminology.
ICH Q9(R1) is a draft revision of the document first produced on 9 November 2005, and currently open for public comment as this revision (R1) until 15 March 2022.
Within the pharmaceutical industry, the principles and framework of ICH Q9, coupled with the official ICH training material that supports this guideline, are instrumental in enhancing the application of effective quality risk management by industry and regulators. As previously stated on many occasions, quality systems are a fundamental requirement in the pharmaceutical industry, and it is evident that quality risk management is a valuable component of an effective quality system.
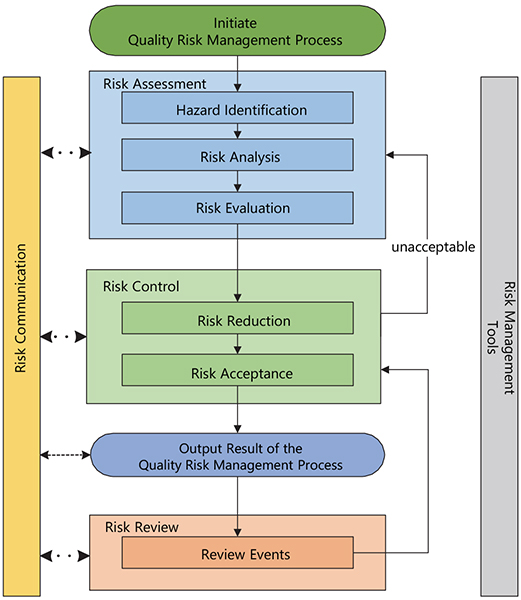
Figure 1. Risk Management cycle. Redrawn from ICH Q9(R1).3 © European Medicines Agency, 2021
If we customise the previously described ISO interested parties/stakeholder statement in relation to pharmaceuticals—although there are a variety of stakeholders, including patients and medical practitioners as well as government and industry—the protection of the patient by managing the risk to quality and availability, when availability risks arise from quality/manufacturing issues, should be considered of prime importance.
The manufacturing and use of a drug (medicinal) product, including its components, necessarily entail some degree of risk. The risk to its quality is just one component of the overall risk. It is important to understand that product quality is assured based on appropriate risk-based decision-making throughout the product lifecycle, such that the attributes that are important to the quality of the drug (medicinal) product are maintained and the product remains safe and effective. An effective quality risk management approach can further ensure the high quality of the drug (medicinal) product to the patient by providing a proactive means to identify and control potential quality issues during development and manufacturing. A proactive approach to quality risk management facilitates continual improvement and is of strategic importance in achieving an effective pharmaceutical quality system. Additionally, use of quality risk management can improve the decision making if a quality problem arises.
In the development phase, quality risk management is part of building knowledge and understanding risk scenarios, so that appropriate risk control can be decided upon during technology transfer, for use during the commercial manufacturing phase. In this context, knowledge is used to make informed risk-based decisions, trigger re-evaluations and stimulate continual improvements.
Effective and proactive quality risk management can facilitate better, more informed and timely decisions throughout the lifecycle. This can provide regulators with greater assurance of a company’s ability to deal with potential risks and avert problems and can beneficially affect the extent and level of direct regulatory oversight.
The application of digitisation and emerging technologies in the manufacture and control of medicinal products can present certain challenges. The application of quality risk management to the design, validation and technology transfer of advanced production processes and analytical methods, advanced data analysis methods and computerised systems is important.
In this revision of ICH Q9 the following paragraph uses the term “lifecycle”, as discussed below.
The purpose of this document is to offer a systematic approach to quality risk management for better, more informed, and timely decisions. It serves as a foundation or resource document that is independent of, yet supports, other ICH Quality documents and complements existing quality practices, requirements, standards, and guidelines within the pharmaceutical industry and regulatory environment. It specifically provides guidance on the principles and some of the tools of quality risk management that can enable more effective and consistent risk-based decisions, both by regulators and industry, regarding the quality of drug substances and drug (medicinal) products across the product lifecycle. It is not intended to create any new expectations beyond the current regulatory requirements. An understanding of formality in quality risk management may lead to resources being used more efficiently, where lower risk issues are dealt with via less formal means, freeing up resources for managing higher risk issues and more complex problems that may require increased levels of rigour and effort. An understanding of formality can also support risk-based decision-making, where the level of formality that is applied may reflect the degree of importance of the decision, as well as the level of uncertainty, complexity and criticality which may be present. Appropriate use of quality risk management can facilitate but does not obviate industry’s obligation to comply with regulatory requirements and does not replace appropriate communications between industry and regulators. Quality risk management should not be used in a manner where decisions are made that justify a practice that would otherwise, in accordance with official guidance and/or regulations, be deemed unacceptable.
Lifecycle Management
During the 2015–2020 cycle, within the Expert Committee structure of USP, an Expert Panel was formed to investigate the extension of PAT and QbD principles into the analytical arena. The output from this Analytical Procedure Life Cycle Expert Panel resulted in the following Stimuli articles, published on this topic in the Pharmacopeial Forum.
- Lifecycle Management of Analytical Procedures: Method Development, Procedure Performance Qualification, and Procedure Performance Verification5
- Fitness for Use: Decision Rules and Target Measurement Uncertainty6
- Analytical Target Probe: Structure and Application Throughout the Analytical Lifecycle7
- Analytical Control Strategy8
- Proposed New USP General Chapter: the Analytical Procedure Lifecycle <1220>9
This set of Stimuli articles summarised the Lifecycle approach and its implementation in the Analytical arena, culminating in the final version of the new USP General Chapter <1220> covering this topic published in Pharmacopeial Forum.10
Significantly, and to bring this discussion up to date, USP General Chapter <1220> has been processed through the USP voting and review process and becomes an official chapter on 1 May 2022.
A synopsis of this new chapter is as follows:
<1220> Analytical Procedure Life Cycle. This new chapter presents an enhanced framework for analytical procedures that holistically incorporates all the events that take place over the procedure lifecycle that are designed to demonstrate that a procedure is, and remains, fit for the intended purpose.
Validation of an analytical procedure is defined in Validation of Compendial Procedures <1225> as “the process by which it is established, by laboratory studies, that the performance characteristics of the procedure meet the requirements for the intended analytical applications”. Generally regarded as an event not a sequence of events or activities being part of a journey from development to retirement.
International Council for Harmonisation guideline ICH Q2(R1) similarly defines “the objective of validation of an analytical procedure is to demonstrate that it is suitable for its intended purpose”.
Considering these definitions more broadly, all activities that confirm that a procedure is suitable for the intended purpose that take place over the entire life of the procedure can be considered under the validation umbrella. Some of these activities are currently described in Transfer of Analytical Procedures <1224>, <1225> and Verification of Compendial Procedures <1226>, which provide guidance for formal validation, transfer and verification of analytical procedures. These are important activities that provide assurance that an analytical procedure is suitable for the intended purpose in the laboratory where the procedures will be used. However, they are often treated as discrete activities that are completed at certain points in the life of a procedure and may not be considered holistically. As such, these events can be disconnected from each other and from other activities that are intended to confirm the procedure remains fit for the intended purpose. These other activities include establishing and ensuring adherence to procedure controls such as system suitability, implementing an appropriate replication strategy for samples and standards and monitoring the procedure during routine use to ensure it continues to provide reportable values that meet the requirements for the measurement. The proposed new chapter seeks to build a framework to link these connections through the lifecycle approach.
The concept of the analytical procedure lifecycle is not entirely new. Some components are described in the chapters mentioned previously, as well as Statistical Tools for Procedure Validation <1210>, and the concept is aligned with quality by design concepts.
The evolutionary process of this whole area of interest, is shown by this statement in the <1220> stimuli article:
The Analytical Procedure Life Cycle Expert Panel acknowledged that some of the life cycle concepts may evolve as ICH Q14 guidance is developed and ICH Q12 guidance is implemented. General chapter <1220> will be re-evaluated for content and terminology when ICH Q14 guidance is finalized and aligned where possible. In the interim, this chapter provides a general framework that encompasses the entire procedure life cycle that can be applied where practical.
Having established the new “Lifecycle” chapter, in the 2020–2025 cycle the newly formed Analytical Instrument and System Qualification Joint Subcommittee (AISQ JSC), risen as the phoenix from the ashes of the Life Cycle Expert Panel, turned its attention to expanding the approach to the Qualification of Analytical systems, and has recently published on 3 January 2020, a Stimuli article titled: “Analytical Instrument and System (AIS) Qualification, to support Analytical Procedure Validation over the Life Cycle” the Abstract summary of which is stated below:
As previously stated in this series of articles, all analytical instruments, and systems in regulated environments, need to be qualified to be “fit for intended use”. In the USP, this is achieved using the information provided in the currently official general chapter Analytical Instrument Qualification 1058. Qualifying the operation and performance of an analytical instrument/system is a critical part of a robust quality management system and is required in a current good manufacturing practice (GMP) environment. In pharmacopeial applications, the performance of an instrument or system directly impacts the data reported by establishing the reportable value of an analytical test characteristic specified in a monograph. For this reason, USP’s Analytical Instrument and System Qualification Joint Subcommittee (AISQ JSC) is considering not only possible enhancements to 1058, but also the impacts on analytical instruments chapters below 1000, as well as other general chapters. As an example, Figure 1 (see Reference 11) illustrates the nature of these interdependencies for a number of the currently official spectroscopic general chapters.
The primary purpose of this Stimuli to the Revision Process (SRP) article, is to share the JSC’s current thinking on AISQ within the USP–NF with all stakeholders, and to gain input on potential revisions and enhancements to 1058. The discussion is focused on the spectroscopic general chapters series 85x and 185x. The JSC intends to publish two additional SRPs, where the second will present the topic of Target Measurement Uncertainty (TMU), which is outlined later in the SRP article, and the third will expound in more detail the qualification cycle of Analytical Instrument and Systems (AIS).
4th Generation: from 2021 forward
As we have seen in this article, expansion of the Quality process has evolved and continues to date into this array of both newly defined, and completely new concepts, such as Analytical Target Profile (ATP), “Fitness for Purpose”, “Proof of Control”, Data Integrity etc.
Please be assured that any significant developments in these areas will be reported in future articles within Quality Matters by the author(s)—so watch this space!
Within the body of the article are also key references to “digitisation of data”, computer systems etc. and, of course, directly related to these terms is the software used to control the systems and produce the data. Therefore, logically, this fundamental component of modern analytical systems, i.e. software and data integrity, will be reviewed in the next article in this series.
References
- ISO/IEC 17025, General Requirements for the Competence of Testing and Calibration Laboratories. International Organization for Standardization (ISO), Geneva, Switzerland (2017).
- ISO 31000:2018, Risk Management. International Organization for Standardization (ISO), Geneva, Switzerland (2018). https://www.iso.org/standard/65694.html
- ICH, ICH guideline Q9 (R1) on Quality Risk Management. European Medicines Agency (2021). https://www.ema.europa.eu/en/documents/scientific-guideline/draft-international-conference-harmonisation-technical-requirements-registration-pharmaceuticals_en-1.pdf
- Risk Management: ISO 31000. International Organization for Standardization (ISO), Geneva, Switzerland (2018). https://www.iso.org/publication/PUB100426.html
- “Lifecycle management of analytical procedures: method development, procedure performance qualification, and procedure performance verification”, Pharmacopeial Forum 39(5) (2013). https://www.uspnf.com/pharmacopeial-forum/pf-legacy-pdfs-archive-28-44
- “Fitness for use: decision rules and target measurement uncertainty”, Pharmacopeial Forum 42(2) (2016). https://www.uspnf.com/pharmacopeial-forum/pf-legacy-pdfs-archive-28-44
- “Analytical target probe: structure and application throughout the analytical lifecycle”, Pharmacopeial Forum 42(5) (2016). https://www.uspnf.com/pharmacopeial-forum/pf-legacy-pdfs-archive-28-44
- “Analytical control strategy”, Pharmacopeial Forum 42(5) (2016). https://www.uspnf.com/pharmacopeial-forum/pf-legacy-pdfs-archive-28-44
- “Proposed new USP general chapter: the analytical procedure lifecycle <1220>”, Pharmacopeial Forum 43(1) (2017). https://www.uspnf.com/pharmacopeial-forum/pf-legacy-pdfs-archive-28-44
- “<1220> Analytical Procedure Life Cycle”, Pharmacopeial Forum 46(5) (2020). https://online.usppf.com/usppf/document/GUID-35D7E47E-65E5-49B7-B4CC-4D96FA230821_10101_en-US
- C. Burgess, M.L.J. Weitzel, J.-M. Roussel, O. Quattrocchi, J. Ermer, R. Slabicky, G.P. Marting and G. Vivó-Truyols, “Analytical Instrument and System (AIS) qualification, to support analytical procedure validation over the life cycle”, Pharmacopeial Forum 48(1) (2022). https://online.usppf.com/usppf/document/GUID-8A30D2D5-7056-460D-A7B0-E93446F9979B_10101_en-US

John Hammond
John Hammond is an experienced analytical scientist, spectroscopist and technical marketing professional, skilled in the development, production and marketing of analytical systems into highly regulated and controlled industries. A Fellow of the Royal Society of Chemistry (FRSC), executive member of ISO/TC334 and an Expert Advisor to the United States Pharmacopeia, General Chapters, Chemical Analysis committee.
[email protected]
